Dongwook Namgung and Young-Chul Choi*
Department of Neurology, College of Medicine, Gangnam Severance Hospital, Yonsei University, Seoul, Republic of Korea
*Corresponding Author:
Young-Chul Choi
Department of Neurology, College of Medicine
Gangnam Severance Hospital, Yonsei University
211 Eonju-ro, Gangnam-gu, Seoul, Republic of Korea
Tel: + 82-2-2019-3320
E-mail: ycchoi@yuhs.ac
Received Date: September 10, 2018; Accepted Date: September 14, 2018; Published Date: September 17, 2018
Citation: Namgung D, Choi YC (2018) A Family with Clinical Phenotypic Variability of X-linked Myotubular Myopathy. Ann Clin Lab Res Vol.6 No. 3:252. DOI: 10.21767/2386-5180.100252
Commentary
X-linked myotubular myopathy (XLMTM) is a form of centronuclear myopathy (CNM) associated with myotubularin. Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in skeletal muscle cells. In CNM the nuclei are located at a position in the centre of the cell, instead of their normal location at the periphery. XLMTM, which is caused by mutations in the MTM1 gene located on Xq28, is considered the most severe form of myotubular myopathy (MTM). XLMTM typically presents with profound muscle weakness, marked hypotonia, and respiratory failure at birth, often leading to death in the first year of life. However, milder phenotypes of XLMTM have been reported [1-3]. Herein, we report a Korean family with XLMTM presenting phenotypic variability including neonatal death and survival into adulthood.
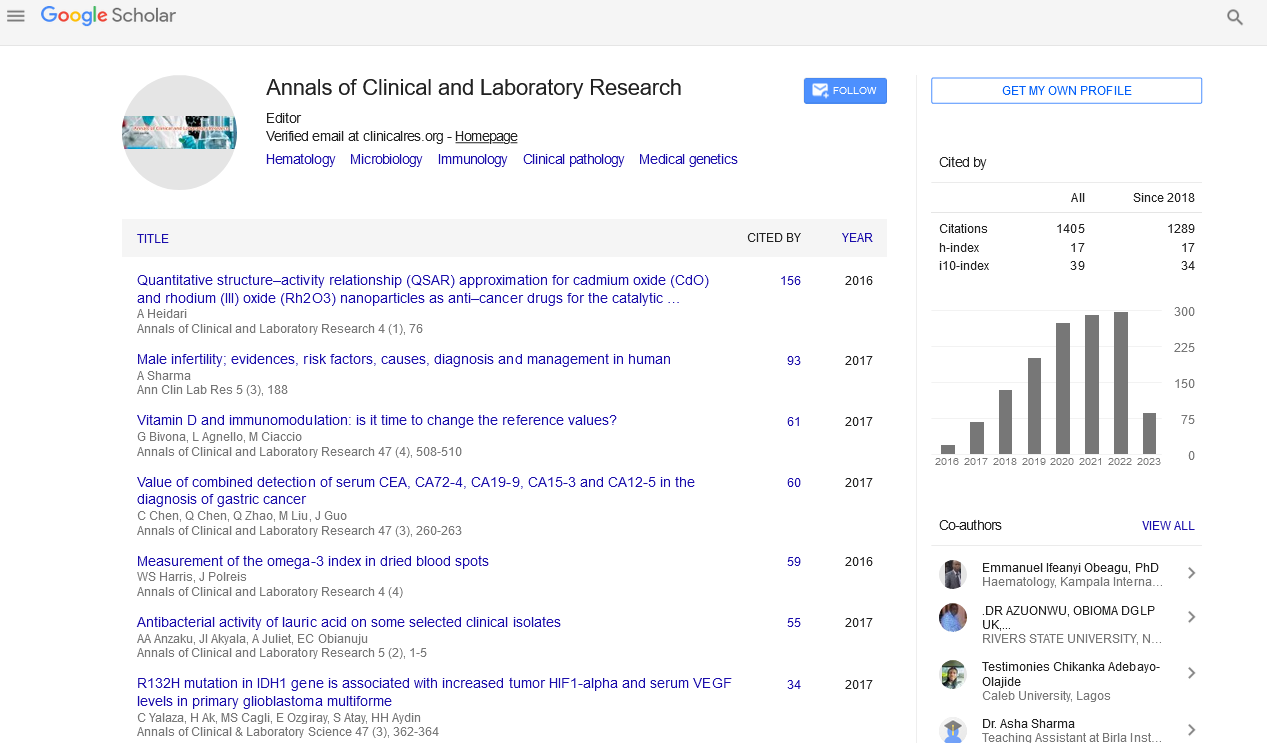
The proband, a 16-year-old boy (Figure 1, III-10), presented to our clinic with general weakness and respiratory difficulty. At birth, he showed weak crying and sucking with severe hypotonia. At 3.5 months after birth, he underwent a muscle biopsy for generalized hypotonia and was diagnosed with congenital myopathy with fibre type disproportion. He started walking at 18 months, but his symptoms had been worsening slowly. When we examined him at the age of 16 years, he showed an elongated face, extra ocular muscle (EOM) weakness, dysarthria, dysphagia, and proximal dominant limb weakness (Medical Research Council (MRC) grade II) with low vital capacity. Laboratory findings showed a normal CK level, but an electro diagnostic study revealed myopathic change. Echocardiography showed an ejection fraction of 35% and dilated cardiomyopathy (DCMP). An overnight non-invasive ventilator was applied.

Figure 1: Pedigree and sequencing chromatograms in a family with X-linked myotubular myopathy. Pedigree of the family (asterisks [*], individuals with genotyping confirmation; arrow, proband; filled, affected; unfilled, unaffected; inner filled circle in large circle, carrier female; small filled circle, spontaneous abortion).
The family history showed that his younger brother (a 10- year-old boy, III-11), cousin (III-3), and uncles (II-1 and II-2) on the mother’s side were affected. His younger brother showed mild hypotonia and sucking weakness at birth, but he was still able to walk, run slowly, and do relatively vigorous physical activities. Neurologic examination showed an elongated face, mild EOM weakness, and proximal dominant limb weakness (MRC grade IV). Laboratory findings and echocardiogram were normal. However, his cousin (III-3) and uncles (II-1 and II-2) had experienced neonatal death due to severe hypotonia and respiratory difficulty. His mother had pectus carinatum and strabismus but showed no abnormal neurological deficit.
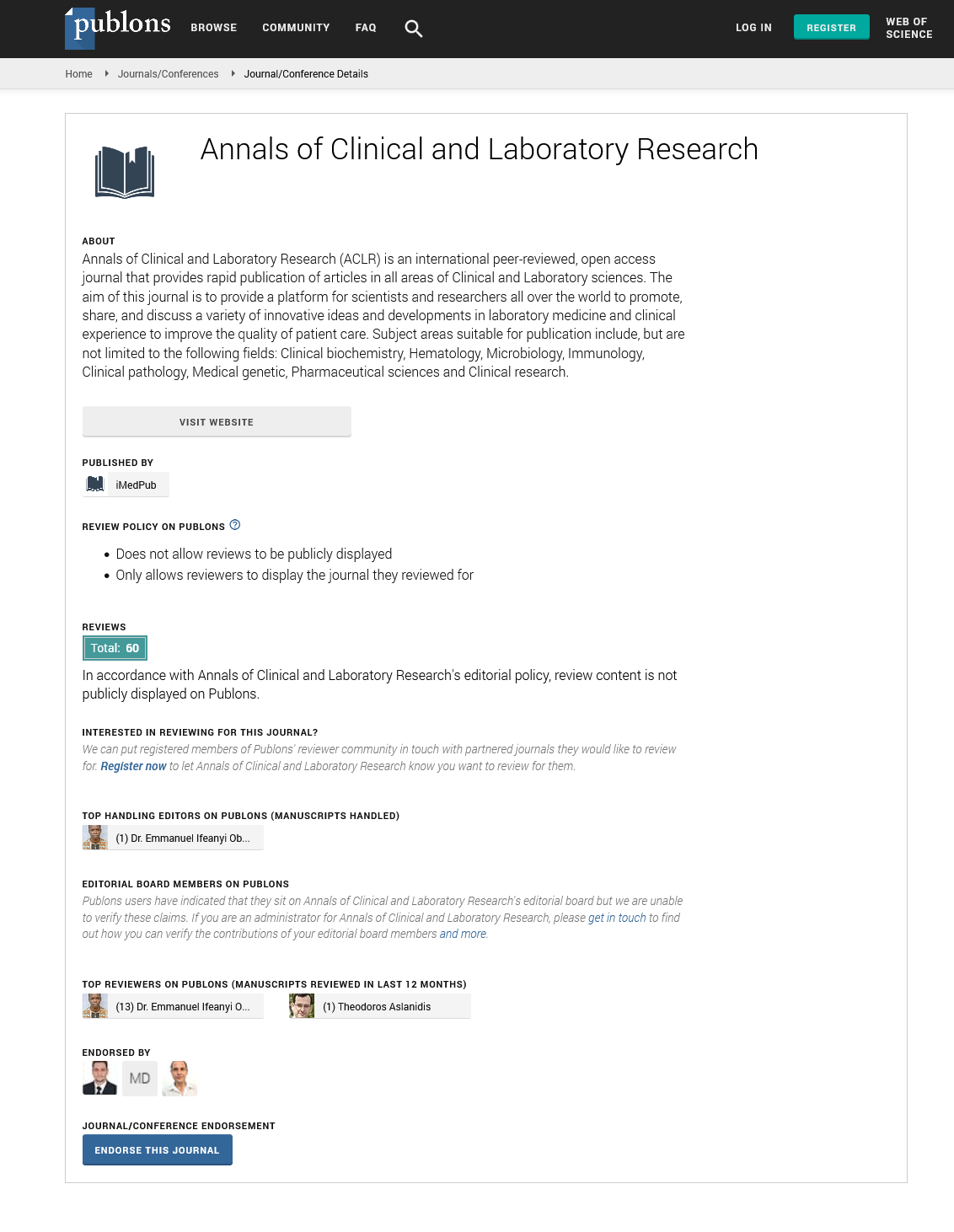
We performed targeted next-generation sequencing on the proband (III-10) and found a hemizygous missense mutation, c. 679G>A (p.V227M), in the MTM1 gene (nomenclature refers to NM_000252.2) (Figure 2). Sanger sequencing of samples from extended family members showed that the younger brother had the same mutation and the mother was a carrier. XLMTM is inherited in an X-linked recessive pattern. In rare cases, females who have one altered copy of the MTM1 gene experience some mild muscle weakness.

Figure 2: Sequencing chromatograms of the MTM1 mutation, c.679G>A (p.V227M), in the proband and younger brother (hemizygote). Arrow indicates the site of the mutation.
To date, about 200 different mutations have been identified in the MTM1 gene [1]. While almost all truncating or splice site mutations showed severe phenotypes, missense mutations of a single amino acid were associated with milder phenotypes [2]. The present missense mutation, c.679G>A (p.V227M) in exon 8, was reported previously as only a mild phenotype with no respiratory support [2,3]. However, this family had variable clinical phenotypes including mild proximal limb weakness (III-11), respiratory difficulty with DCMP (III-10), and neonatal death (II-1, II-2, and III-3). These findings demonstrate phenotypic variability for this missense mutation even in the same family.
The MTM1 gene consists of 15 exons and provides instructions for producing an enzyme called myotubularin. Myotubularin is thought to be involved in the development and maintenance of muscle cells. This enzyme acts as a phosphatase, which means that it removes clusters of oxygen and phosphorus atoms (phosphate groups) from other molecules. Myotubularin removes phosphate groups from two molecules called phosphatidylinositol 3-phosphate and phosphatidylinositol 3,5-biphosphate. These molecules are found within cell membranes and are likely involved in transporting molecules within cells. The myotubularin protein contains consensus sequences for the phosphatase active site in exon 11 and an SET (Suvar3-9, Enhancer-of-zeste, Trithorax) interaction domain in exons 12 and 13. These domains are essential for the function of myotubularin, and missense mutations affecting these domains are associated with a severe phenotype [3]. However, the present mutation and other missense mutations, such as I225T in exon 8 and R241C in exon 9, also present with a severe phenotype, which implies that other uncharacterized protein domains are important for the stability of myotubularin [3].
In conclusion, this report revealed phenotypic variability with both prolonged survival and severe neonatal forms of missense mutation (p.V227M) in the MTM1 gene in the same family. This finding will be important for genetic counselling about prognosis in XLMTM. Although mild forms of XLMTM have long-term survival, monitoring cardiopulmonary function is necessary for management. A future study is recommended to investigate the effects of missense mutations on the function of myotubularin and phenotype correlation.
Conflict of Interest
The authors declare no conflict of interests.
23303
References
- Biancalana V, Caron O, Gallati S, Baas F, Kress W, et al. (2003) Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet 112: 135-142.
- Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, et al. (2000) MTM1 mutations in X-linked myotubular myopathy. Hum Mutat 15: 393-409.
- Herman GE, Kopacz K, Zhao W, Mills PL, Metzenberg A, et al. (2002) Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum Mutat 19: 114-121.