Keywords
mRNA; Vaccine; Immunotherapy; Cancer vaccines
Introduction
Several studies have demonstrated the immunogenicity of tumors, therefore, cancer cells may potentially be recognized and attacked by immune system, and melanoma is one of the best-defined immunogenic tumor [1-4].
Essential components of the mammalian immune system are the antigen presenting cells (APCs), a heterogenous group of immune cells specialized to process and presenting antigens for recognition by certain lymphocytes such as T-cells[5,6]. Therefore, they represent a vital component of the innate immunity and play a pivotal role in the generation and regulation of adaptive immunity by cell-cell interaction (e.g. APC-T-cell interaction) and release of regulatory cytokines (e.g. IL-12, IL-18 and IFN -α or -β) [7,8]. Classical APCs include dendritic cells (DCs), macrophages and B-cells, although DCs are the most powerful APC of the mammalian immune system [9]. Represent a key link between innate and adaptive immunity, given their innate immunity features. Hence, DCs are an optimal target in the development of anti-microbial or anti-cancer vaccines.
Essentially, it is possible to resume the innate properties of DCs in the following sentinel and sensor roles in the immune system: (i) Capture, process and present G antigens (ii) Migrate into lymphoid organs, where naïve T-cells are retained, and (iii) Rapid differentiation or maturation and production of immune enhancing cytokines, in response to several stimuli ranging from innate immunity receptors (e.g. Toll-like receptors, TLRs) [10,11]. Pattern recognition receptors (PRRs) represent the sentinels of the innate immune system, expressed essentially onto the surface of dendritic cells and macrophages and they are able to recognize specific pathogen associated molecular patterns (PAMPs) [12].
The family of TLRs is the class of PRRs that it has been studied most extensively and represent an important group of receptors, as previous studies demonstrated that innate immune system activation can be used as vaccine adjuvants to potentiate the adaptive immune response due their ability to activate various immune cells simultaneously, through the release of specific cytokines [13,14].
The use of nucleic acid-based vaccines (DNA or RNA encoding for an antigen) is a very attractive strategy, as can induce efficiently both humoral and cellular immune responses, which makes tumor escape less likely [15,16]. Additionally, nucleic acidbased vaccine oppositely to more classic and diffused vaccine formulation containing protein or peptide antigens, offer several advantages: (i) Unlike peptide-based vaccines, they do not require prior knowledge; (ii) They are not restricted by the patient’s HLA type; (iii) Finally, nucleic acids can act as adjuvant by providing costimulatory signals, for example, via toll-like receptors (TLRs) [16-19]. The nucleic acid vaccines consist of the delivery of DNA or RNA, encoding for a specific antigen, into the APCs; using one of the following strategies: (i) ex-vivo transfection of APCs; or (ii) in vivo APC-targeting.
Despite the promising features of DNA vaccines, in general, they have been found to elicit a relative lack immune response compare to RNA-based vaccines [19-22]. The reasons for this are not completely clear, but possible explanations include inefficient delivery of DNA into human cells, due to the fact that DNA needs to cross both cell and nuclear membranes to be transcribed, the low expression of DNA-sensing machinery, and the differing expression of nucleic acid sensing PRRs [17,18]. The relatively poor immunogenicity of DNA vaccines, combined with concerns about their potential for oncogenesis via integration into the host genome, has led to a shift from DNA vaccines towards RNA vaccines.
In the past, several viral vectors were proposed as DCtransfection systems for the delivery of antigen-mRNA, but these kinds of vectors shown some limitations [23]. First, a poorly understood but well-documented immunological phenomenon called immunodominance may occur, whereby other antigenic proteins, such as viral antigens, mask or suppress the response to the less potent tumour antigens [24]. Second, activated Cytotoxic T lymphocyte (CTLs) may recognize and kill antigen-presenting DCs, because DCs expressing both viral and tumour antigens, therefore DCs can be eliminated by virus-specific T cells before they can activate a tumour-specific response [25]. If a viral vector is used to deliver the tumour antigen, this elimination effect would be amplified with subsequent immunizations, as it is likely that the virus-specific T-cell pool would be expanded with each vaccination. Hence, it seems desirable to develop methods of DC transfection that lead to the expression of only those proteins toward which an immune response is desired.
Non-viral gene delivery systems were introduced as alternative to viral-based systems. These vectors have many advantages, such as easy of fabrication and low immune response [26]. The biggest disadvantage of non-viral vectors in clinical use is their low in vivo transfection efficiency. But, contrarily to the classic gene therapy approaches, for gene-based immunotherapy a relatively low transfection efficiency can be sufficient to elicit a strong humoral and cell-mediated antigen-specific immune response and promote tumor regression and the generation of memory that can prevent relapse.
cell/tissue targeting is not requested, because it was demonstrated that DC targeting and activation is possible by simple systemic delivery of the mRNA-loaded carrier, without the need for ligand functionalization [27]. Recently, non-viral vectors have been widely explored as vehicles for mRNA delivery in the area of mRNA-based immunotherapy, but their clinical application is limited, because usually, an elevated dose of mRNA is needed for antigen-specific immunization [27-30].
Here we propose a self-assembled PEG-PAsp (DET) (polyethylene glycol-b-poly{N’-[N-(2-aminoethyl)-2-aminoethyl] aspartamide}) block copolymer/ PAsp (DET) (poly{N’-[N-(2-aminoethyl)-2- aminoethyl] aspartamide}) homopolymer polyplex as efficient platform for mRNA-based vaccine (Figure 1A). Although this polymer has been widely explored as an alternative for gene delivery vector, it has never been used as platform for nucleic acid-based vaccination. The ability of the proposed system to induce a specific immunization was confirmed employing an mRNA encoding for Ovalbumin (OVA) chicken protein, an antigen commonly used as a model protein for studying antigen-specific immune responses in mice. Interestingly, we show our vaccine formulation has an intrinsic self-adjuvant propriety given its ability to lead, in the transfected DCs, a robust release of IL-12 and IFN-β, which are recognized as a potent vaccine adjuvant, by TLR 7/8 pathway activation. Moreover, OVA-mRNA-loaded nanovector was able to induce a high cell-mediated and humoral antigen-specific immune response.
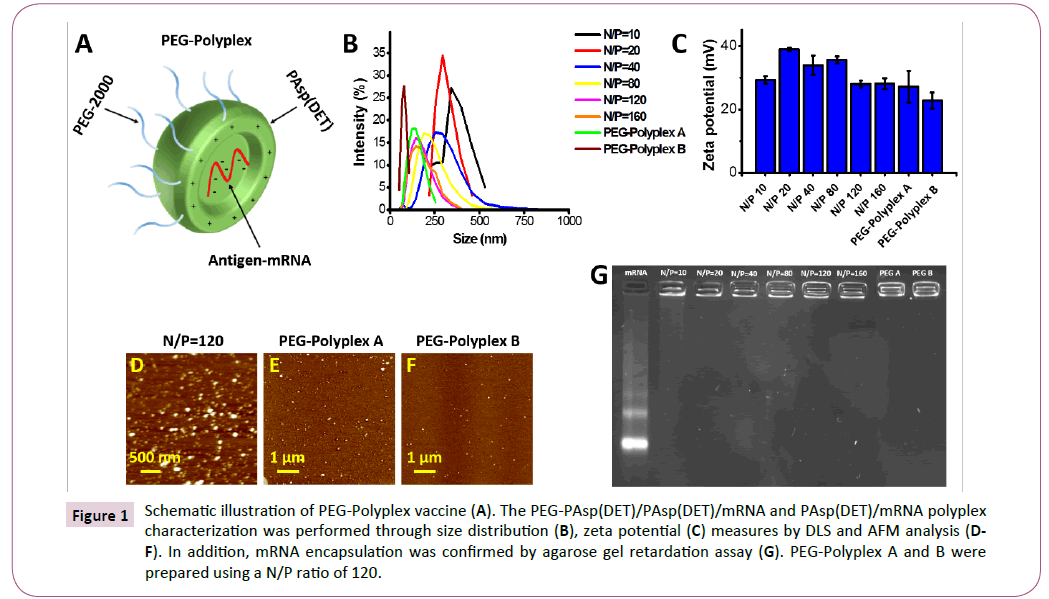
Figure 1: Schematic illustration of PEG-Polyplex vaccine (A). The PEG-PAsp(DET)/PAsp(DET)/mRNA and PAsp(DET)/mRNA polyplex characterization was performed through size distribution (B), zeta potential (C) measures by DLS and AFM analysis (DF). In addition, mRNA encapsulation was confirmed by agarose gel retardation assay (G). PEG-Polyplex A and B were prepared using a N/P ratio of 120.
Finally, a murine OVA-B16 lung metastatic melanoma model was employed to investigate the anti-tumor effect of the antigenmRNA loaded polyplex vaccine. Immunization of B16-OVAbearing mice with OVA-mRNA loaded Polyplex significantly reduced up to 93% the number of lung metastases.
Material and Methods
Polyplex (PAsp (DET)/mRNA) and PEGPolyplex (PEG-PAsp (DET)/PAsp (DET)/ mRNA) preparation and characterization
PEG-PAsp(DET) block polymer, MW of PEG~12,000 Da, DP of PAsp(DET) ~70 and PAsp(DET) homopolymer with DP of ca. 50, were purchased from NOFAmerica corporation. One volume of PAsp(DET) and/or PEG-PAsp(DET) solution (PAsp(DET)/PEGPAsp( DET) mole ratio of 100:1 and 50:1, for PEG-polyplex A and B respectively) was mixed with two volume of mRNA solution in 10 mM Tris-HCl buffer (pH 7.4) to a final volume of 90 μl. The Polyplexes were prepared using different N/P ratio (amine group on PAsp (DET) and phosphate groups on RNA). After incubation at room temperature for 30 minutes, the complexes are ready to use. The size distribution and zeta potential were measured using a Malvern Zetasizer Nano ZS dynamic light scattering (DLS) instrument (Malvern Instruments, Worcestershire, UK). The mRNA (1 mg/mL) was purchased from Trilink Biotechnologies (San Diego, CA).
Gel retardation assay
Polyplexes and PEG-Polyplexes (250 ng of eGFP-mRNA) were loaded with 1 x RNA loading buffer (BioRad, Hercules, CA) and run on a 0.7% agarose gel in standard 1 x TBE buffer (BioRad, Hercules, CA) at 75 mV for 1 hour. The RNA bands were stained with GelRed nucleic acid gel stain (Biotium, Hayward, CA) and then detected by a GelDoc system (BioRad, Hercules, CA).
Atomic Force Microscopy (AFM)
Silicon substrate was treated with piranha solution (H2SO4/H2O2, 1:2 (vol/vol)) for 1 hour at 80°C and washed with distilled water thoroughly and blow-dry. Then, the substrate was functionalized with 2-(carbomethoxy) ethyltrimethoxysilane (CETES). Briefly, the silane was diluted to a final concentration of 2% in 95% Isopropyl Alcohol (IPA) (made freshly by adding 5% water to 100% IPA). Then the substrate was submerged in the silane solution and incubated under shaking in a Thermomixer at 1400 rpm for 2 hours at 35°C. The substrate was washed 3 times Rinse with 100% IPA and blow-dry. Then, 20 μl of diluted polyplexes solution (1:100 or 1:200) was dropped onto the silanes-silicon surface and incubated for 20 minutes. After the surface was washed 3 times with distilled water and the substrate was immediately transferred onto the AFM stage to be analyzed.
Stability of polyplexes against heparin
Polyplexes were prepared and incubated for 30 min with heparin solutions of different concentrations, expressed as heparin/ mRNA (w/w) ratio (1, 2, 3, 4 and 5). The samples were run on a 0.7% agarose gel 1 x TBE buffer (BioRad, Hercules, CA) at 75 mV for 1 hour. RNA bands were stained with GelRed nucleic acid gel stain and then detected by a GelDoc system.
Generation of bone marrow derived cells (BMDCs)
Dendritic cells from C57BL/6 mice were obtained from bone marrow as previously described [31]. Briefly, BMDCs from femur and tibia were flushed out using 1% FBS-containing PBS using a 5-ml syringe. The cells were treated with ACK lysis buffer (Lonza Inc.) to remove red blood cells and centrifuged at 500 g for 4 minutes. The BMDCs were resuspended in RPMI-1640 culture medium supplemented with 10% FBS (Atlas Biological, Fort Collins, CO), 0.1% β-mercaptoethanol, 1% penicillin/streptomycin (Pen/Strep, 10,000 units of penicillin and 10 mg of streptomycin, Sigma-Aldrich, Saint Louis, MO). The cytokines Granulocyte- Macrophage Colony-Stimulating Factor (GM-CSF) (20 ng ⁄ ml) and IL-4 (20 ng ⁄ ml) were added and cells were set at a density of 1 x 10 6cells ⁄ ml. The medium was changed every two days and non-adherent cells were harvested as immature BMDCs. The cells were used for all the transfection experiments at day 5. The dendritic cells have been routinely tested for CD11c expression and ≥85% of cells are positive for this marker.
In vitro transfection using an eGFP-mRNA
BMDCs were seeded in a 24-well plate at density of 3 x 105in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS, Atlas Biological, Fort Collins, CO), 1% streptomycin/ Penicillin (Sigma-Aldrich) and 0.1% β-mercaptoethanol (Sigma- Aldrich). After 24 hours of incubation, the dendritic cells were incubated for with PEG-Polyplexes prepared using different N/P ratios (80, 120 and 160) and loaded with 0.5 μg eGFPmRNA. Fluorescence images were captured using a fluorescent microscopy (Eclipse TE2000-S microscope Nikon Corporation, Tokyo, Japan). Flow cytometry analysis was used to quantify the transfection and translation efficiency in BMDCs. Cells were harvested and resuspended in PBS with 2% FBS and cytometric analysis was performed using a BD Accuri C6 flow cytometer (Becton Dickinson, BD, Franklin Lakes, NJ, USA). Gates were set to exclude necrotic cells and cellular debris and the fluorescence intensity of events within the gated regions was quantified. Data were collected on 20,000 events for each sample.
Cytotoxicity test by MTS assay
Cytotoxicity assays were performed using tetrazolium compound based CellTiter 96® AQueous One Solution Cell Proliferation (MTS) assay (Promega). To test for cellular cytotoxicity induced by polyplexes, the cells were cultured in 96-well plate, with appropriated complete media (Final volume 200 μl) at a concentration of 104 cells/well. Then, 0.1 μg mRNA loaded polyplexes were added and MTS assay was then performed according to the manufacturer’s instruction at 24 hours after treatment.
Cellular uptake mechanism
DC2.4 cells (ATCC) were seeded in 24-well plates at a density of 1.5 x 105 cells/ml and incubated for 24 h. Then, they were treated with PEG-Polyplexes preloaded with eGFP mRNA after 30 minutes of incubation with different inhibitors such as amiloride (0.2 mM), chloroquine (100 mM), genistein (50 μm), chlorpromazine (15 μm) pimozide (10 μm). The cells were washed with icecold PBS and the mean fluorescence intensity was observed by fluorescent microscopy (Eclipse TE2000-S microscope Nikon Corporation, Tokyo, Japan).
Evaluation of dendritic cells maturation markers by flow cytometry analysis
Dendritic cell activation was achieved by in vitro transfection using OVA-mRNA. DC2.4 cells were seeded in 24-well plates at a density of 1.5 × 105 cell/well in 1 ml of RPMI complete medium (10% FBS, 1% Pen/Strep, 0.1% β-mercaptoethanol) 24 h prior to transfection. Then, PEG-polyplexes containing 0.5 μg of mRNA were incubated with DC2.4 cells at 37°C for 24 hours. After 24 hours, the cells were washed with PBS and stained for CD11c, CD40, CD86 and MHC II. The cells were analyzed by BD Accuri C6 flow cytometer (Becton Dickinson, BD, Franklin Lakes, NJ, USA). All the antibodies used for this experiment were purchased from BD Bioscience.
OVA-antigen presentation by dendritic cells
BMDCs were transfected with PEG-polyplexes loaded with 0.5 μg of OVA-mRNA, and after 48 hours the cells were stained for 10 minutes at room temperature with 25-D1.16 monoclonal antibody (MHC class I H-2Kb, BD Bioscience). Subsequently, the cells were stained 30 minutes with an anti-CD11c (BD Bioscience) and then evaluated using a BD Accuri C6 flow cytometer (Becton Dickinson, BD, Franklin Lakes, NJ, USA).
MHC I and II–restricted antigen presentation assays
BMDCs were seeded at a concentration of 3 x 105 cells/ml in a 24- well plate and incubated for 24 hr at 37°C in RPMI-1640 medium (10% FBS, 1% Pen/Strep and 0.1% β-mercaptoethanol). Then, at day 5 of culture, the dendritic cells were transfected with PEGPolyplexes loaded with 0.5 μg of OVA-mRNA. After 48 hours, the transfected dendritic cells were washed and co-cultured with B3Z, T–T hybridomas against OVA (257–264)/H-2Kb complex, and DOBW, T–T hybridoma against OVA (323–339)/I-Ad complex, T-cells using a T-cell/dendritic cells ratio of 1:1. T-cell activation was monitored at 24 hours by measuring IL-2 accumulation in the supernatant by ELISA (eBioscience). Data shown are from triplicate experiments.
Innate immunity activation
DC2.4 cells were cultured in 24-well plates at a density of 1.5 x 105 cells in 1 ml of complete medium in the presence of PEGPolyplexes loaded with 0.5 μg OVA-mRNA. LPS was used at 100 ng/ml as a positive control. After 24 hours incubation, the supernatants were collected, then IL-6, TNF-α, IFN-β and IL-12 concentrations were measured using sandwich ELISA kit (R&D Systems, Minneapolis, MN).
TLR inhibitors
TLR7 (ODN 2087) and TLR7/8 (ODN 20959) inhibitors were purchased from Militenly Biotec (San Diego, CA). DC2.4 cells were seeded in a 24-well plate at a density of 1.5 x 105 in 1 ml of RPMI-1640 complete medium and incubated 24 hours at 37°C. Then, the dendritic cells were pre-incubated for 1 hour at 37 with 2.5 μm of TLR inhibitor. Subsequently, PEG-Polyplexes loaded with 0.5 μg of OVA-mRNA were added into the wells and after 24 hours the culture medium was collected to be analysed for IL-12 and IFN-β levels by ELISA kit. The dendritic cells without pre-incubation with TLR inhibitor were used as positive control, whereas the dendritic cells incubated in absence of OVA-mRNA loaded PEG-Polyplexes with/without inhibitor represent the negative controls.
Mice immunization by two dose injections of OVA-mRNA loaded polyplexes
PEG-Polyplex and Polyplex vectors were prepared using an N/P ratio of 120 following the procedure described above. Then, two groups of five BALB/c mice were injected s.c. with polyplexes and PEG-Polyplexes loaded with 2 μg of OVA-mRNA. A second s.c. injection was performed after 3 days from the first administration. Control mice were injected in the same manner with PBS. The mice were sacrificed 7 days after the first injection to allow spleen, lymph nodes, and blood collection. Protamine sulfate/mRNA polyplexes were prepared, mixing protamine sulfate (Sigma Aldrich) and OVA-mRNA (Trilink Biotechnologies) at an w/w ratio of 2 in 10 mM Tris-HCl buffer, pH 7.4, followed by 20 minutes of incubation at room temperature.
Ex-vivo detection of antigen-specific T-cells by ELISPOT
Spleens and lymph nodes were harvested from immunized and control groups of mice. Single-cell suspensions were obtained and blood cells were lysed using ACK buffer and the cell suspensions passed through a 70 μm cell strainer (BD Falcon). To quantify the IFN-γ production by ex-vivo stimulated antigenspecific T-cells, cells were plated in RPMI-1640 medium at a density of 3 x 104 cells/well in UNIFILTER® 96-well ELISpot plates. Consecutively, cells were stimulated with 2 ng/ml OVA peptides (OVA257-264, OT-I, and OVA323-339, OT-II) and OVA protein. Following overnight incubation (15–18 hr) at 37°C, the cells were washed 3 times with PBS containing 0.05% Tween-20. Plates were incubated for 3 to 4 hr at room temperature with biotinylated mouse secondary antibody for IFN-γ (BioLegend). After 3 washes, as before, the plates were further incubated for 2 hr with antirabbit IgG HRPO (SIGMA) and then processed for staining using an AEC kit according to manufacturer's instructions (Vector Laboratories). The number of spots forming cells was analyzed using ImmunoSpot software (Cellular Technology).
Measure ex-vivo IFN-γ release by ELISA assay
T-cells were isolated from spleen and lymph nodes as previously described. The collected cells were cultured in 24-well plates at a density of 1 x 106 cells/ml and incubated with 2 ng/ml of OVA peptides (OVA257-264 specific for MHC class I and OVA323-339, specific for MHC class II) and OVA protein at 37°C. After 24 h incubation, the culture supernatant was harvested and the presence of IFN-γ was tested using a commercial mouse IFN-γ immunoassay ELISA kit (BD Bioscience) according to the manufacturer's instructions. The concentrations of IFN-γ in the samples were determined from the standard curves.
Measure of OVA-antibodies titters
OVA-specific antibodies titters were measured in the serum of immunized mice. Blood was collected by retro-orbital bleeding on day 14 after immunization, and allowed to clot overnight at 4°C. The serum was separated by centrifugation at 1000 x g for 20 min. Serum was collected and anti-OVA antibody titters were determined by enzyme-linked immunosorbent assay, as previously described. Briefly, 96-well plates (Nunc-Immuno) were coated by overnight incubation at 4°C with 100 μl/well of PBS containing OVA at 40 μg/ml. Plates were blocked with 1% bovine serum albumin in PBS for 2 h, and serial twofold dilutions of serum samples in PBS were added to the wells. After a 2-h incubation, plates were then washed with PBS containing 0.05% Tween 20 and incubated for 1 h at room temperature with HRPconjugated goat anti-mouse immunoglobulin G (IgG), IgG1, IgG2c, IgG2b and Immunoglobulin M (IgM) antibodies (Southern Biotechnology Associates). After three additional washes, the plates were then incubated with tetramethylbenzidine substrate. The reaction was stopped by the addition of 1N HCl (Sigma). Absorbance was read at 450 nm with a microplate reader (Bio- Rad). Ab Titers were calculated as the inverse dilution at which the absorbance equaled that of control mice plus 2 standard deviations.
In vivo toxicity of PEG-Polyplex
Cytokine analysis was performed to assess the cytokine response inducing by PEG-Polyplex exposure. A MAP Mouse Cytokine/ Chemokine Magnetic Bead Panel - Premixed 32 Plex - Immunology Multiplex Assay (EMD Millipore, Darmstadt, Germany) was used to measure the cytokine levels such as IL-1b, IL-2, IL-6, IL-12(p70), TNF-α, IFN-γ, and RANTES. BALB/c mice were s.c. injected with 2 μg OVA-mRNA loaded micelles and after 24 hours serum samples were analyzed in duplicate wells according to the manufacture's instruction.
In vitro CD8+ T-cell mediated anti-cancer activity
DC2.4 cells were seeded at a density of 1.5 × 105 cells/well in 24-well plate with 1 ml of RPMI-1640 medium. After overnight incubation, dendritic cells were pre-incubated with TLR7 (ODN 20958, Miltenyi Biotec, Germany) and TLR7/8 (ODN 2087, Miltenyi Biotec, Germany) inhibitors for 1 hour at 37°C. Then, 0.5 μg of OVA-mRNA loaded pegylated-polyplex were added and incubated for 24 hours at 37°C. Subsequently, OVA-specific CD8+ T-cells (B3Z cells) were co-cultured with the transfected DC2.4 at T-cell/DC2.4 ratio of 2:1 and incubated together for 24 hours at 37°C. B16-OVA tumor cells were plated in a 96-well tissue culture plate at 0.5 × 104 cells per well (200 μl) and incubated overnight at 37°C. The tumor cells were co-cultured with activated B3Z cells at effector/target ratio of 5:1 for 4, 6 and 24 hours at 37°C. The viability of tumor cells was then determined using an MTS formazan viability assay (Promega, Madison, WI). The optical density of the wells was read at 490 nm on a plate reader. The mean viability of the treated tumor cells was calculated as a percentage relative to the control wells with non-treated cancer cells (100% survival). All the samples were measured in triplicate.
Metastatic lung cancer model
Mice were injected with 2.5 × 105 B16-OVA melanoma tumor cells intravenously to establish lung metastases [32]. Three days later, tumour-bearing mice were s.c. vaccinated with 2 μg OVA-mRNA loaded micelles. Mice were boosted at days 7 and 10. Lungs were harvested 18 days following tumor inoculation and fixed with 4% of paraformaldehyde. The lung metastases were counted under a dissecting microscope.
TRP2-specific CD8+ T-cells detection in vivo
C57BL/6 mice were immunized s.c. with TRP2-mRNA loaded PEG-Polyplex at day 3, 7 and 10 using the same dose of TRP2- mRNA (2 μg). One week after the last immunization the blood was collected and analyzed by flow cytometry for the presence of tetramer positive CD8+ T-cells (H-2b/TRP2 tetramer, MBL). Tetramer positive cells are expressed as the percentage of CD3+/ CD8+ T cells.
Statistical analysis
Comparisons between experimental groups were made by using the two-tailed Student t-test. P < 0.05 was considered significant.
Results and Discussion
Characterization of mRNA-loaded polyplexes
Size and zeta potential of the polyplexes were evaluated by dynamic light scattering (DLS) measurements (Figures 1B and 1C), which showed smaller cumulant diameter and a lower polydispersity index in the pegylated-polyplexes (PEG-Polyplex) compared with non-pegylated polyplexes (Polyplex). The polyplexes size decrease with increasing of N/P ratio in the range from 10 to 80, and beyond this range the size distribution remain constant. But a further size reduction was observed in response to the pegylation and this variation is proportionated to the PEG-PAsp (DET)/PAsp(DET) ratio used for polyplex preparation. Higher zeta potential was measured with N/P ratio between 20- 80 and over this range, the superficial charge of the polyplex remain positive although, decrease significantly. Moreover, it is evident that the inclusion of PEG-PAsp(DET) block copolymer, in the formulation, induce a relevant reduction of the zeta potential (Table 1).
| Polyplex |
Z-Average(nm) |
PDI (Polydispersity index) |
| N/P=10 |
1298.3 |
0.643 |
| N/P=20 |
737.3 |
0.568 |
| N/P=40 |
329.6 |
0.335 |
| N/P=80 |
231.4 |
0.267 |
| N/P=120 |
158.4 |
0.259 |
| N/P=160 |
157.7 |
0.228 |
| PEG-Polyplex |
Z-Average (nm) |
PDI (Polydispersity index) |
| Formulation A |
147.9 |
0.224 |
| Formulation B |
96.45 |
0.186 |
Table 1: Average size and polydispersity index (PDI) for the size intensity distribution plots shown in Figure 1B. The polyplexes were prepared using an eGFP-mRNA (996 nt).
Size and morphology of the non-pegylated and pegylatedpolyplexes were confirmed by atomic force microscopy (AFM), which confirmed a size decrease in response to the pegylation and that the size variation is proportioned to the amount of block copolymer included in the polyplex formulation (Figures 1D-1F). Whereas, the incorporation of mRNA into the polyplex was confirmed by gel retardation assay (Figure 1G). The gel retardation assay revealed complete mRNA incorporation into the polyplexes already employing an N/P ratio of 20 (Figure 1G).
Serum and extracellular matrix proteins can lead to vector disassembly [33]. To evaluate the possible effects of anionic proteins on PEG-Polyplex stability, we tested the susceptibility of our Nano vector to heparin displacement. Heparin is one of the glycosaminoglycans (GAG), which are negatively charged polysaccharides that are the major components of the extracellular matrix in many tissues and it is also present on the cell surface [34]. Polyplexes were incubated with increasing heparin concentrations. Polyplex dissociation in terms of mRNA release was shown by electrophoresis of the samples on an gelred-containing agarose gel (Figure 2). Any dissociation did not occur even at high heparin/mRNA (w/w) ratios.

Figure 2: Heparin displacement assay PEG-Polyplexes were prepared at N/P ratio of 120 and incubated for 30 min at 37°C in the presence of increasing amounts of heparin, expressed as heparin/siRNA (w/w) ratio.
In vitro optimization of transfection conditions
Primary cells are known to be resistant to transfection, therefore, after optimized the polyplex formulation, we decided to evaluate the transfection efficiency of eGFP-mRNA-loaded polyplexes with primary dendritic cells, which represent our target to promote an antigen-specific immune response. BMDCs (Bone marrowderived cells) were obtained from femurs of BALB/c mice and cultured with granulocyte macrophage-colony stimulating factor (GM-CSF) and IL-4 to induce dendritic cells differentiation.
The dendritic cells at day five were transfected with polyplexes loaded with 0.5 μg of eGFP-mRNA and 48 hours post-transfection GFP-positive cells were detected by fluorescence microscopy (Figures 3A-3E). Three different polyplex formulation, obtained using an N/P ratio of 80, 120 and 160, were tested for transfection ability with dendritic cells. The transfection efficiency was measured by FACS analysis and a higher number of GFP-positive cells were observed using an N/P ratio of 120 (Figure 3F). Importantly, MTS assay did not detect any significant cytotoxicity, therefore as previously demonstrated PAsp (DET) polymer has an optimal biocompatibility (Figure 3G) [35,36].
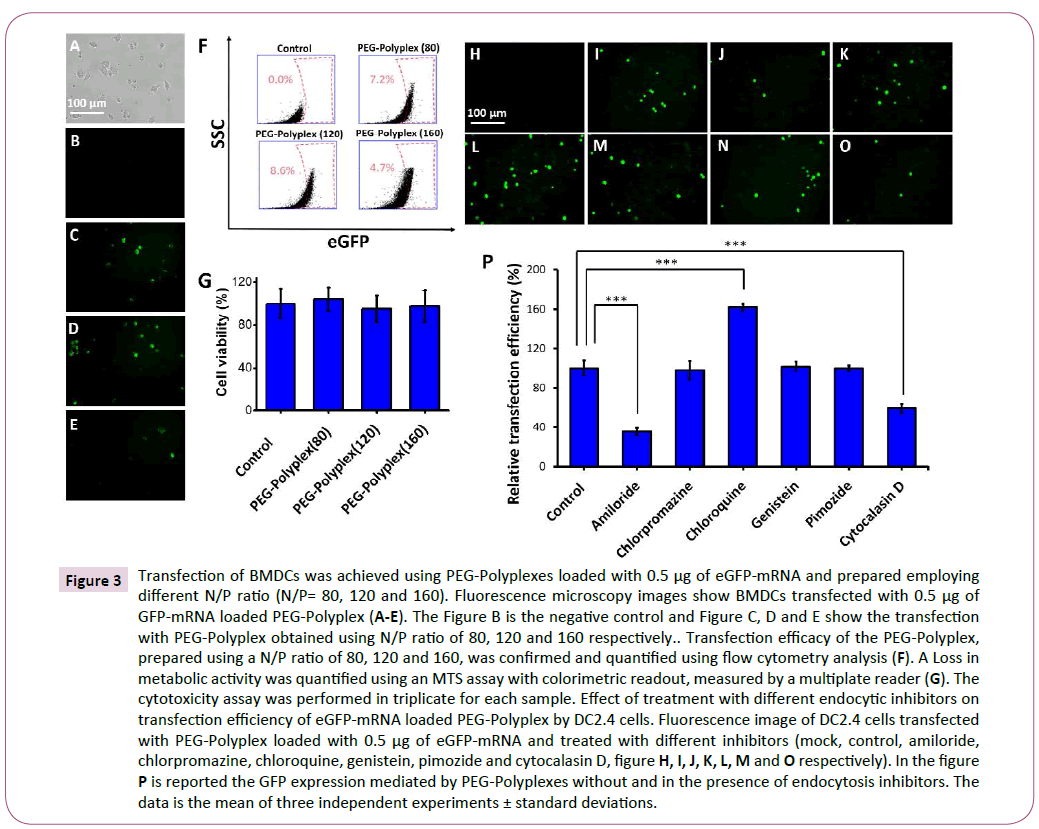
Figure 3: Transfection of BMDCs was achieved using PEG-Polyplexes loaded with 0.5 μg of eGFP-mRNA and prepared employing different N/P ratio (N/P= 80, 120 and 160). Fluorescence microscopy images show BMDCs transfected with 0.5 μg of GFP-mRNA loaded PEG-Polyplex (A-E). The Figure B is the negative control and Figure C, D and E show the transfection with PEG-Polyplex obtained using N/P ratio of 80, 120 and 160 respectively.. Transfection efficacy of the PEG-Polyplex, prepared using a N/P ratio of 80, 120 and 160, was confirmed and quantified using flow cytometry analysis (F). A Loss in metabolic activity was quantified using an MTS assay with colorimetric readout, measured by a multiplate reader (G). The cytotoxicity assay was performed in triplicate for each sample. Effect of treatment with different endocytic inhibitors on transfection efficiency of eGFP-mRNA loaded PEG-Polyplex by DC2.4 cells. Fluorescence image of DC2.4 cells transfected with PEG-Polyplex loaded with 0.5 μg of eGFP-mRNA and treated with different inhibitors (mock, control, amiloride, chlorpromazine, chloroquine, genistein, pimozide and cytocalasin D, figure H, I, J, K, L, M and O respectively). In the figure P is reported the GFP expression mediated by PEG-Polyplexes without and in the presence of endocytosis inhibitors. The data is the mean of three independent experiments ± standard deviations.
After having confirmed that polyplexes efficiently deliver the mRNA into the DCs, we wanted to elucidate the mechanism by which this cells uptake mRNA-loaded polyplexes. Therefore, we examined the different cellular uptake pathways for the polyplexes using various endocytic inhibitors; each one specific for a particular endocytic pathway (Figures 3H-3P). The amiloride (blocks macropinocytosis by Na+/H+ exchanger inhibition) significantly decreased the cellular transfection by 70% (Figure 3P), proving that our polyplexes are able to release efficiently the mRNA into the cytosol mainly exploiting micropinocytosis pathway. A prominent role of micropinocytosis in the PEG-Polyplex entry into dendritic cells was confirmed by the substantial reduction (~45%) in transfection efficiency following pre-treatment with cytochalasin D an inhibitor of actin polymerization (Figure 3P).
Moreover, this study has revealed that adding genistein (inhibits caveolin-mediated endocytosis), chlorpromazine (inhibits clathrin-mediated endocytosis), Pimozide (inhibits phagocytosis) did not significantly altered the cellular transfection efficiency. On the contrary, chloroquine, known to prevent endosome acidification and maturation, induced a 2-fold improved transfection efficiency. Still, it has not been clarified the mechanism by which chloroquine increase the transfection efficiency, but it seems likely that endolysosomal accumulation of chloroquine inhibit acidification by proton-buffering which induce to increase the osmotic pressure within the endosome that lead to endosomal membrane disruption with the consequent antigen escape into the cytosol, enabling its translation and processing.
In vitro validation of PEG-Polyplex for mRNAbased vaccine applications
With the ability to deliver antigen-mRNA into the cytosol of dendritic cells, we then determined that this property could be combined to elicit dendritic cells maturation. Dendritic cells were transfected with 0.5 μg of mRNA encoding for Ovalbumin (OVAmRNA), a chicken protein used as a model antigen for studying antigen-specific immune responses in mice. 48 hours posttreatment dendritic cells displayed a shift from the immature to the mature phenotype through upregulation of the costimulatory molecules CD40, CD86, and MHCII (Figure 4A).
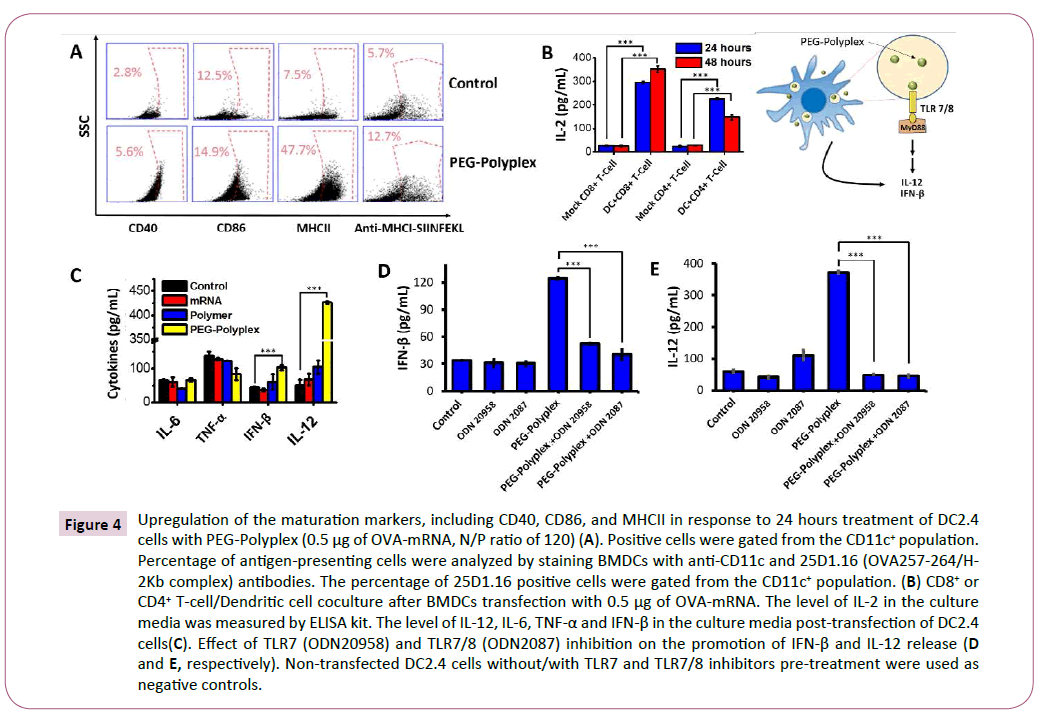
Figure 4: Upregulation of the maturation markers, including CD40, CD86, and MHCII in response to 24 hours treatment of DC2.4 cells with PEG-Polyplex (0.5 μg of OVA-mRNA, N/P ratio of 120) (A). Positive cells were gated from the CD11c+ population. Percentage of antigen-presenting cells were analyzed by staining BMDCs with anti-CD11c and 25D1.16 (OVA257-264/H-2Kb complex) antibodies. The percentage of 25D1.16 positive cells were gated from the CD11c+ population. (B) CD8+ or CD4+ T-cell/Dendritic cell coculture after BMDCs transfection with 0.5 μg of OVA-mRNA. The level of IL-2 in the culture media was measured by ELISA kit. The level of IL-12, IL-6, TNF-α and IFN-β in the culture media post-transfection of DC2.4 cells(C). Effect of TLR7 (ODN20958) and TLR7/8 (ODN2087) inhibition on the promotion of IFN-β and IL-12 release (D and E, respectively). Non-transfected DC2.4 cells without/with TLR7 and TLR7/8 inhibitors pre-treatment were used as negative controls.
MHC class II molecules are crucial for the activation of CD4+ T cells, which is well known for their critical role in the rising and regulation of adaptive anti-tumour response. T cells can directly mediate cytotoxicity against tumor cells.
To test the ability of activated DCs to process antigen the presence of dendritic cells presenting OVA257– 264 peptide (SIINFEKL) on the major histocompatibility complex I (MHC-I) was analyzed 48 hours after treatment. The mRNA-based vaccine was capable of generating a significant number of SIINFEKL-MHC-I+ DCs (Figure 4A).
Primed-T-cells secrete several cytokines, such as interlukin-2 (IL-2), which influence various lymphocyte subsets during the immune responses. For example, IL-2 has a crucial role in the maintenance of regulatory T (Treg) cells and for the differentiation of CD4+ T cells into specific effector T-cell subsets in response to antigen-mediated activation [37]. Furthermore, IL-2 signal for CD8+ T-cells regulate both effector T-cell generation and differentiation into memory cells [38]. The elevated levels of IL-2 measured in the medium, using an ELISA assay after dendritic cells-T cells co-culture confirm the ability of the dendritic cells to activate T-cells by MHC-antigen presentation after stimulation with OVA-mRNA-loaded polyplex (Figure 4B). An important advantage of mRNA-based immunotherapies over traditional recombinant protein vaccines is the ability to activate innate immunity by TLR signalling. TLRs were found act as adjuvant receptors, that promote and potentiate adaptive immune system by stimulating the release of several cytokines, including type I interferon and IL-12. Type I interferon and IL-12 are recognized as a powerful adjuvant that potentiate the antigen-specific immune response, because capable of activating T- and NK- cells for IFN-γ production and to promote CD+ Th1 immunity [38,39]. Immune deviation towards Th1 responses results in tumor rejection as Th1 pathways typically produce activation of cytotoxic T-cell lymphocytes (CTL), natural killer (NK) cells, macrophages and monocytes, all of which can attack cancer cells and generally defend against tumors. A significant up-regulation of IFN-β (1.7 fold) and a robust secretion of IL-12 (8 fold) were observed after treatment of DCs with OVA-mRNA-loaded polyplexes. Whereas, a non-significant change in the levels of TNF-α and IL-6 was detected in response to the treatment with OVA-mRNA-loaded polyplexes. Interestingly, naked-mRNA and PAsp (DET) polymer were not able to induce IFN-β and IL-12 release (Figure 4C). The role of TLRs in the promotion of IL-12 and Type I interferon secretion, by OVA-mRNA-loaded polyplexes treated DCs, was proved using TLR7 (ODN20958) and TLR7/8 inhibitors (ODN2087). The release of IL-12 and IFN-β is significantly compromised by the inhibition of TLR7/8 pathway (Figures 4D and 4E). Therefore, we can conclude that mRNA-loaded polyplexes induce innate immune system activation by TLR7/8 signalling.
In vivo antigen-specific immunization by administration of OVA-mRNA loaded PEGpolyplexes
To assess the success of in vivo immunization in response to s.c. injection of OVA-mRNA-loaded polyplexes, we determined the presence of antigen-specific IFN-γ secreting T-cells in the spleen and lymph nodes of treated mice. The mice were immunized by subcutaneously (s.c.) injection of 2 μg OVA-mRNA, boosted at day 3 with the same dose and, spleens and lymph nodes were harvested at day 7. The presence of OVA-, OTI-, OTII- specific IFN-γ secreting T-cells was confirmed by ELISA and ELIspot assays (Figures 5A-5D). High levels of IFN-γ and significant number of antigen-specific IFN-γ secreting T-cells were detected both in the spleen (Figures 5A and 5B) and lymph nodes of immunized mice, confirming the ability of the proposed vaccine formulation to promote an antigen-specific adaptive immune response.
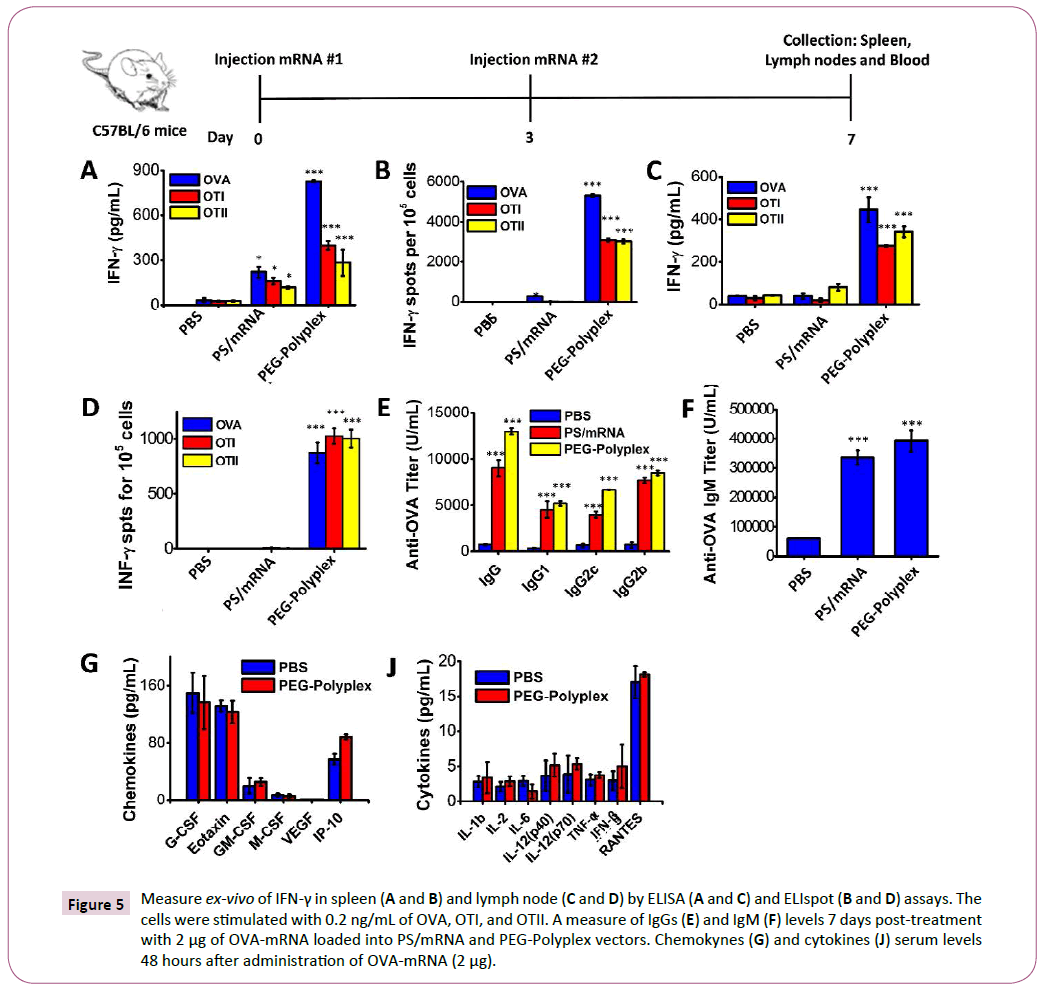
Figure 5: Measure ex-vivo of IFN-γ in spleen (A and B) and lymph node (C and D) by ELISA (A and C) and ELIspot (B and D) assays. The cells were stimulated with 0.2 ng/mL of OVA, OTI, and OTII. A measure of IgGs (E) and IgM (F) levels 7 days post-treatment with 2 μg of OVA-mRNA loaded into PS/mRNA and PEG-Polyplex vectors. Chemokynes (G) and cytokines (J) serum levels 48 hours after administration of OVA-mRNA (2 μg).
Induction of OVA-specific immunoglobulin G (IgG, IgG1, IgG2b and IgG2c) and IgM were detected in the serum at day 7 after first OVA-mRNA vaccine administration (Figures 5E and 5F). Given the high levels of OVA-specific IgG and IgM detected in the serum of the vaccinated mice, we can affirm that the proposed vaccine efficiently induces a potent antigen-specific humoral response. Moreover, s.c. injection induced high OVA-specific IgG1 and IgG2a titers using only a small amount of OVA-mRNA (2 μg), demonstrating that the mRNA-based immunotherapy promotes both Th1 and Th2 type of antibody-mediated immunity.
With the purpose to demonstrate that the proposed systems represent an important advancement of the current state-ofthe- art in the area of mRNA-base vaccine implementation, we decided to compare our vaccine formulation against protamine sulfate/mRNA polyplex (PS/mRNA), a non-viral vector widely employed for antigen-mRNA delivery and currently under study in several clinical trials for anti-cancer immunotherapy [40-42]. The detection of OVA-specific IFN-γ secreting cells in the spleen and lymph nodes of vaccinated mice, revealed a significantly higher number of primed T-cell in the mice treated with OVAmRNA loaded PEG-Polyplex than mice vaccinated with PS/OVAmRNA polyplex. Contrarily, equivalent titers of IgG and IgM, it was found in both vaccination with OVA-mRNA loaded PEGPolyplex and with PS/OVA-mRNA complex.
A major barrier to the clinical application of nanomaterials is their potential toxicity. Therefore, we assessed the toxicity associated with OVA-mRNA-loaded polyplex using wild-type mice. A 32-plex Luminex assay was employed to measure cytokine and chemokine serum levels, including IL-1b, IL-2, IL-6, IL-12(p40), IL-12(p70), TNF-α, IFN-γ, RANTES, Eotaxin G-CSF, GMCSF, M-CSF, VEGF, and IP10. As reported in the Figures 5G and 5J, 24 hours post-treatment we can exclude any polyplex-related pro-inflammatory or toxicity.
Anticancer effect of OVA-mRNA loaded PEGPolyplex in vitro and in vivo
To measure the antigen-specific cell-mediated immune response promoted by immunization with OVA-mRNA-loaded PEGPolyplex vaccine, we employed an in vitro Cytotoxic T Lymphocyte (CTL) cytotoxicity assay. OVA-mRNA transfected DCs were coincubated with OVA-specific CD8+ T-cells using a ratio of 1:2, then primed-T cells were co-culture with B16-OVA tumor cells at effector/target cells ratio of 5:1. CD8+ T-cell-mediated antitumor activity was estimated measuring tumor cell viability at 4, 6 and 24 hours by MTS assay (Figure 6A). Using TLR7/8 inhibitors, we proved the concept that inhibition of this pathway, with the consequent block of type-I IFN and IL-12 secretion, affect the anti-tumour cytotoxic immune response. Indeed, it is well known that IL-12 enhances the function and anti-tumour activity in murine and human CD8+ T-cells [43].
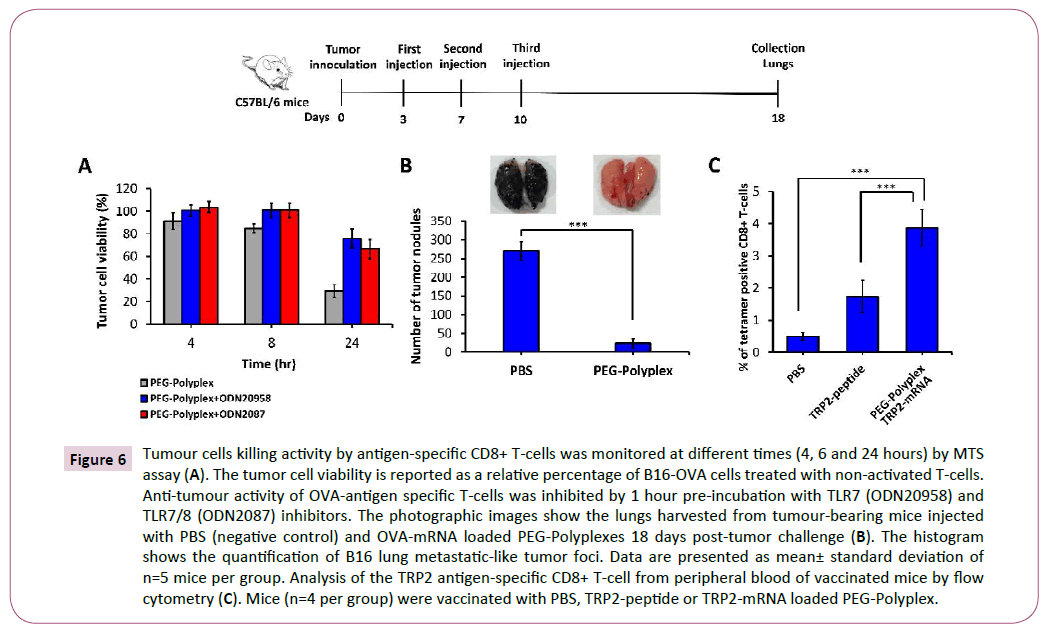
Figure 6: Tumour cells killing activity by antigen-specific CD8+ T-cells was monitored at different times (4, 6 and 24 hours) by MTS assay (A). The tumor cell viability is reported as a relative percentage of B16-OVA cells treated with non-activated T-cells. Anti-tumour activity of OVA-antigen specific T-cells was inhibited by 1 hour pre-incubation with TLR7 (ODN20958) and TLR7/8 (ODN2087) inhibitors. The photographic images show the lungs harvested from tumour-bearing mice injected with PBS (negative control) and OVA-mRNA loaded PEG-Polyplexes 18 days post-tumor challenge (B). The histogram shows the quantification of B16 lung metastatic-like tumor foci. Data are presented as mean± standard deviation of n=5 mice per group. Analysis of the TRP2 antigen-specific CD8+ T-cell from peripheral blood of vaccinated mice by flow cytometry (C). Mice (n=4 per group) were vaccinated with PBS, TRP2-peptide or TRP2-mRNA loaded PEG-Polyplex.
Anti-cancer mRNA-based immunotherapy is able to induce an antigen-specific cell-mediated and humoral immune responses sufficient to obtain tumor regression. To address this question, we generated an OVA-B16 melanoma lung metastasis tumor model and treated it at day 3 through s.c. injection of OVA-mRNAloaded polyplexes, followed by two booster doses at days 7 and 10. The mice were sacrificed at day 18 post-tumor inoculation. As shown in Figure 6B, mice without vaccination (injected with PBS), developed extensive pulmonary metastases. However, a marked reduction in pulmonary metastases (up to 93%) was observed in mice treated with OVA-mRNA-loaded polyplexes (Figure 6B).
Conclusion
In conclusion, we demonstrated, that using a low dose of antigenmRNA, the administration of antigen-mRNA-loaded polyplexes can induce a potent anti-tumor immunity, effective for cancer treatment. Our results contribute to a relatively small number of studies published on mRNA-loaded nanoparticle mediating tumor vaccination. In addition, our findings also support the concept that mRNA-based vaccine offers the advantages to promote simultaneously antigen-specific adaptive cell-mediated, humoral immune response, and it can act as self-adjuvant, promoting innate immunity by TLRs. TLR signalling promotes the release of several cytokines, such as type I interferon and IL-12 that have the capacity to potentiate the anti-tumor adaptive immune response.
Self-assembled are fast and inexpensive to prepare and potentially can be used as a universal platform for anti-tumour mRNAbased immunotherapy approaches, given that any polypeptide can be encoded by mRNA. Thus, the mRNA-loaded polyplex introduced here can represent a novel and valid platform for the development of mRNA-based vaccines.
Acknowledgement
This work was funded by the Italian Flagship Project NanoMax (S.P.)
19913
References
- Lesterhuis WJ, Haanen JB, Punt CJ (2011)Cancer immunotherapy – revisited. Nat Rev Drug Disc 10: 591-600.
- Blankenstein T, Coulie PG, Gilboa E, Elizabeth MJ (2012) The determinants of tumour immunogenicity.Nature Reviews Cancer 12:307-313
- Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptative immune cells in the tumor microenvironment. 14: 1014-1022.
- Maio M (2012) Melanoma as a model tumor for immuno-oncology. Ann Oncol 23: 10-14.
- Janeway CAJ, Travers P, Walport M (2001)Immunobiology: The immune system in health and disease. (5th edn). Principles of innate and adaptive immunity, Garland Science, New York, USA.
- Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nature immunology 14: 1014–1022.
- Friedl P, Den Boer AT,Gunzer M(2005) Tuning immune responses: Diversity and adaptation of the immunological synapse. Nat Rev Immunol 5: 532-545.
- Medzhitov R (2001)Toll-like receptors and innate immunity. Nat Rev Immunol 1: 135-145.
- Stockwin LH, McGonagle D, Martin IG, Blair GE (2000) Dendritic cells: Immunological sentinels with a central role in health and disease. Immunol Cell Biol78: 91–102.
- Martín-Fontecha A, Lanzavecchia A,Sallusto F (2009)Dendriticcell migration to peripheral lymph nodes. HandbExpPharmacol. 188:31-49.
- Alvarez D, Vollmann EH, von Andrian UH(2008) Mechanisms and Consequences of Dendritic Cell Migration. Immunity 29: 325.
- Takeuchi O, Akira S (2010)Pattern recognition receptors and inflammation. Cell 140: 805-820.
- Steinhagen F, Kinjo T, Bode C, Klinman DM(2009)TLR-based immune adjuvants. Vaccine 29: 3341-3355.
- Fotin-Mleczek M, Duchardt KM, Lorenz C, Pfeiffer R, Ojkic-Zrna S, et al. (2011) Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide anti-tumor activity. J Immunother 34: 1-15.
- Vogel FR, Sarver N (1995)Nucleic acid vaccines.Clinical MicrobiolRev 8: 406-410.
- Restifo NP, Ying H, Hwang L, Leitner WW(2000)The promise of nucleic acid vaccines. Gene Ther 7: 89-92.
- Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, et al. (2013) Therapeutic cancer vaccines: Past, present, and future. AdvCan Res 119: 421–475.
- Coban C, Kobiyama K, Aoshi T, Takeshita F, Horii T, et al. (2011) Novel strategies to improve DNA vaccine immunogenicity. Curr Gen Ther11: 479–484.
- Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ (2011) Developing mRNA-vaccine technologies. RNA Biol 9: 1319–1330.
- Ulmer JB, Mason PW, Geall A, Mandl CW (2012) RNA-based vaccines. Vaccine 2:4414-4418.
- Sahin U, Kariko K,Türeci O (2014) mRNA-based therapeutics-developing a new class of drugs. Nat Rev Drug Disc 13: 759–780.
- McNamara MA, Nair SK,Holl EK (2015) RNA-based vaccines in cancer immunotherapy. J ImmunolRes.
- Choi Y, Chang J(2013)Viral vectors for vaccine applications. ClinExp Vaccine Res 2: 97-105.
- Yewdell JW, Bennink JR (1999) Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol 17: 51–88.
- Ingulli E, Mondino A, Khoruts A,Jenkins MK (1997)In vivo detection of dendritic cell antigen presentation to CD4+ T cells. J Exp Med 185: 2133-2141.
- Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, et al. (2014) Non-viral vectors for gene-based therapy. Nat Rev Genet 15: 541–555.
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, et al. (2016) Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534: 396–401.
- Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K et al.(2012)Nonviral delivery of self-amplifying RNA vaccines. ProcNatlAcadSci U S A 109: 14604-14609.
- Broos K, Van der Jeught K, Puttemans J, Goyvaerts C, et al. (2016)Particle-mediated intravenous delivery of antigen mRNA results in strong antigen-specific T-cell responses despite the induction of type I interferon. Nucleic acids 5:e326.
- Pardi N, Hogan MJ,Pelc RS, Muramatsu H (2017) Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543: 248–251.
- Lacombe MH, Hardy MP, Rooney J, Labrecque N(2005)IL-7 receptor expression levels do not identify CD8+ memory T lymphocyte precursors following peptide immunization. J Immunol 175: 4400-4407.
- Overwijk WW, Restifo NP(2001)B16 as a mouse model for human melanoma. CurrProtocImmunolChapter: Unit–20.1.
- Buyens K, Lucas B, Raemdonck K, Braeckmans K, Vercammen J, et al.(2008)A fast and sensitive method for measuring the integrity of siRNA-carrier complexes in full human serum. J Control Release 126: 67–76.
- Sarrazin S, Lamanna WC, Esko JD (2011)Heparansulfate proteoglycans.Cold Spring HarbPerspectBiol 3:a004952.
- Itaka K, Ishii T, Hasegawa Y, Kataoka K(2010) Biodegradable polyamino acid-based polycations as safe and effective gene carrier minimizing cumulative toxicity. Biomaterials 31:3707-14.
- Akagi D, Oba M, Koyama H, Nishiyama N, Fukushima S, et al. (2007)Biocompatible micellarnanovectors achieve efficient gene transfer to vascular lesions without cytotoxicity and thrombus formation. Gene Therapy 14: 1029-1038.
- Boyman O, Sprent J(2012)The role of interleukin-2 during homeostasis and activation of the immune system. NatRev Immunol 12:180-190.
- Bliss J, Van Cleave V, Murray K, Wiencis A, Ketchum M, et al.(1996) IL-12, as an adjuvant, promotes a T helper 1 cell, but does not suppress a T helper 2 cell recall response. J Immunol156: 887-894.
- Proietti E, Bracci L, Puzelli S, Di Pucchio T, Sestili P, et al. (2002) Type I IFN as a natural adjuvant for a protective immune response: Lessons from the influenza vaccine model. J Immunol 169:375-383.
- Weide B, Pascolo S, Scheel B, Derhovanessian E, Pflugfelder A, et al. (2009) Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunothe. 32:498-507.
- Fotin-Mleczek M, Zanzinger K, Heidenreich R, Lorenz C, Thess A, et al. (2012) Highly potent mRNA based cancer vaccines represent an attractive platform for combination therapies supporting an improved therapeutic effect. J Gene Med 14: 428-439.
- Kubler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, et al.(2015)Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J Immunother Cancer 3: 26.
- Rubinstein MP, Su EW, Suriano S, Cloud CA, Andrijauskaite K, et al.(2015)Interleukin-12 enhances the function and anti-tumor activity in murine and human CD8(+) T cells. Cancer ImmunolImmunother 64:539-549.

