Introducción
VPH y Cáncer Cervical
A nivel mundial el cáncer cervical (CaCu) es uno de los tumores más comunes y una de las principales causas de muerte entre mujeres. [1]. En el año 2000 murieron a causa de esta enfermedad más de 200,000 pacientes alrededor del mundo [2]. En algunos países subdesarrollados el CaCu es la principal causa de muerte en pacientes en edad reproductiva y económicamente activas, estos casos están ligados frecuentemente al acceso limitado a un diagnóstico oportuno o a un tratamiento médico efectivo [1].
Desde hace 30 años, algunos autores señalaron que el CaCu presentaba un comportamiento de enfermedad de transmisión sexual [3], mientras que otros evaluaron el posible papel del virus del papiloma humano (VPH) en el desarrollo de esta neoplasia [4,5]. Sin embargo, no fue hasta Noviembre de 1991 que la asociación entre la infección por VPH y CaCu quedó oficialmente establecida, después de haber considerado la evidencia epidemiológica y molecular que el ADN de VPH se encuentra presente en más del 99% de los casos de esta neoplasia [6,7].
Actualmente, se han identificado más de 100 tipos de VPH, de los cuales 40 infectan el epitelio genital [8]. Los VPH genitales son clasificados de acuerdo a su potencial para inducir lesiones cervicales; de tal forma que los virus de bajo riesgo están asociados con las verrugas genitales, mientras que los de alto riesgo tienen el potencial de desarrollar cáncer invasor [9]. Entre los tipos virales de alto riesgo oncogénico, los tipos 16 y 18 son los más frecuentes, con una incidencia de alrededor del 50% para el tipo 16 y del 15% para el 18 [9]. Afortunadamente, no todos los pacientes infectados con VPHs oncogénicos desarrollaran CaCu, debido a la eliminación espontánea de secuencias virales. En diferentes estudios epidemiológicos se ha determinado que aproximadamente el 70% de las lesiones precursoras no progresarán a carcinoma invasor, sino que por el contrario, en un período de seis meses a un año la lesión será eliminada. Estos datos indican que la mayoría de las infecciones con VPH son subclínicas [10,11]. Por lo tanto, la infección con virus de alto riesgo es una causa necesaria pero no suficiente para el desarrollo de CaCu; de este modo, otros factores inherentes al hospedero (celulares, inmunológicos, genéticos, epigenéticos) o al ambiente (dieta, contacto con productos contaminantes, etc) pueden incidir en el desarrollo de la enfermedad. Con respecto a los factores virales que conllevan al desarrollo del CaCU, estos han sido estudiados profundamente, produciendo suficiente evidencia para postular la existencia de tres eventos importantes en el desarrollo de un tumor invasor: la integración del ADN viral al genoma del hospedero, la expresión de las oncoproteínas virales E6 y E7, y finalmente la compleja red de interacciones entre E6/E7 con proteínas celulares. En este modelo de carcinogénesis, la identificación de los factores virales, del hospedero y ambientales que influyen en el riesgo de progresión de la enfermedad-desde lesiones cervicales tempranas a un cáncer invasor-, nos conducirá a un mayor entendimiento de la historia natural de la infección por VPH .
Una vez que el VPH ha infectado las células basales, el genoma viral es replicado activamente como episoma y se expresan los genes tempranos (E1-E7). E1 y E2 son proteínas importantes para la replicación del genoma viral y la terminación del ciclo viral [12]. E1 juega un importante papel en el mantenimiento del genoma viral como episoma [13]. En cambio, E2 esta involucrado en la regulación negativa de la actividad transcripcional de los oncogenes virales E6 y E7 [14]. No obstante, E5, E6, y E7 son requeridos para incrementar la proliferación celular basal conduciendo a un incremento en el índice de replicación del genoma viral; por lo tanto, la expresión limitada de estos genes puede darse durante las primeras etapas de la infección [15]. Los genes tardíos (L1 y L2) codifican proteínas de la cápside viral y se expresan durante las últimas etapas del ensamblaje del virus, en las capas medias y superiores del epitelio. Finalmente, los viriones son encapsulados y liberados dentro del tracto genital [16] en donde, pueden infectar otras áreas del epitelio o ser transmitidos sexualmente (Figura 1).
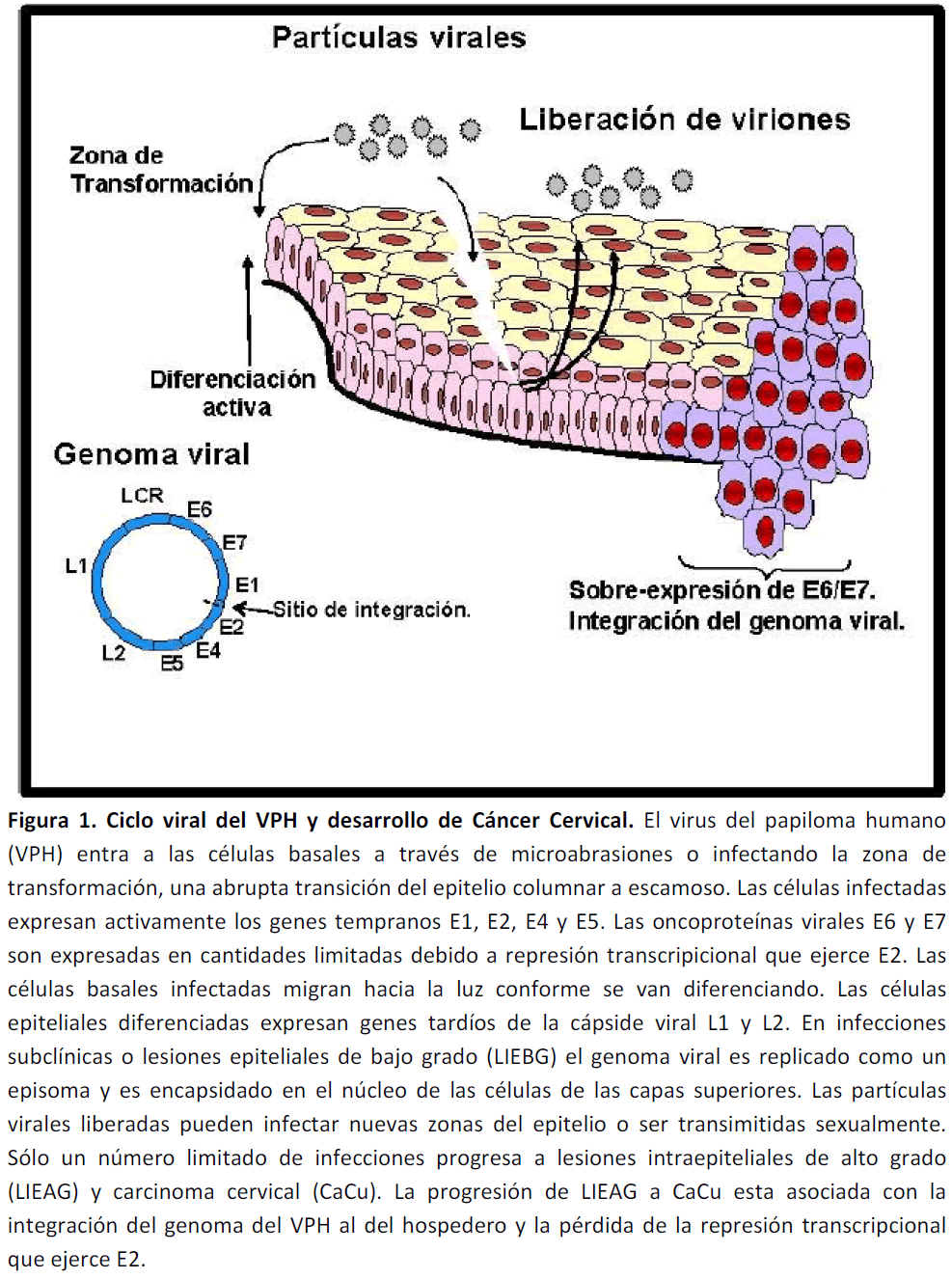
Figura 1. Ciclo viral del VPH y desarrollo de Cáncer Cervical. El virus del papiloma humano (VPH) entra a las células basales a través de microabrasiones o infectando la zona de transformación, una abrupta transición del epitelio columnar a escamoso. Las células infectadas expresan activamente los genes tempranos E1, E2, E4 y E5. Las oncoproteínas virales E6 y E7 son expresadas en cantidades limitadas debido a represión transcripicional que ejerce E2. Las células basales infectadas migran hacia la luz conforme se van diferenciando. Las células epiteliales diferenciadas expresan genes tardíos de la cápside viral L1 y L2. En infecciones subclínicas o lesiones epiteliales de bajo grado (LIEBG) el genoma viral es replicado como un episoma y es encapsidado en el núcleo de las células de las capas superiores. Las partículas virales liberadas pueden infectar nuevas zonas del epitelio o ser transimitidas sexualmente. Sólo un número limitado de infecciones progresa a lesiones intraepiteliales de alto grado (LIEAG) y carcinoma cervical (CaCu). La progresión de LIEAG a CaCu esta asociada con la integración del genoma del VPH al del hospedero y la pérdida de la represión transcripcional que ejerce E2.
Un importante paso durante la infección por VPH corresponde a la integración viral en el genoma del hospedero. El genoma del VPH es replicado usualmente como episoma o molécula extracromosomal en lesiones cervicales precursoras benignas. En tejidos provenientes de tumores, se puede encontrar al ADN viral tanto de forma episomal como integrado [17]. Debido a que el genoma de VPH es una molécula en forma de anillo, requiere de una conformación abierta para que esta se integre al genoma del hospedero; este proceso de rompimiento ocurre frecuentemente en los marcos de lectura abierta E1-E2. Parte de E2 y regiones adyacentes a E2-E4 como E5 y L2 son eliminadas después de la integración; por lo tanto, la represión transcripcional parcial ejercida por E2 se pierde y los oncogenes virales E6 y E7 son expresados activamente en tejidos neoplásicos con genomas virales integrados [18]. Se ha observado que las regiones cromosómicas donde se integra el genoma viral, frecuentemente se encuentran genes involucrados o asociados al proceso de tumorigénesis, como MYC, NR4A2, hTERT, APM-1, FANCC, TNFAIP2, etc [19].
La expresión de E6 y E7 y sus interacciones con proteínas celulares probablemente han sido el centro de la investigación biomédica relacionada al CaCU durante los últimos 20 años. El núcleo central del modelo clásico de E6/E7 es la unión y la inactivación de las proteínas supresoras de tumor p53 y pRb, respectivamente; y fue establecido a finales de 80´s y principios de los 90´s [20,21]. Actualmente, se conoce bien que E6 y E7 interactúan con una gran cantidad de proteínas celulares que participan en vías moleculares involucradas en la activación y establecimiento del fenotipo maligno.
Una de las vías de señalización candidatas que ha sido recientemente estudiada en el modelo de CaCu es la vía de Wnt/ß-catenina. El papel de la señalización de Wnt en cáncer fue descrito inicialmente hace 20 años con el descubrimiento seminal del gen wnt-1 como sitio de integración para el virus de tumor mamario de ratón (MMTV) [22]. Desde entonces una gran cantidad de información ha destacado el papel de la vía de Wnt en el control de varios procesos biológicos, por ejemplo, procesos de diferenciación celular, proliferación, migración, adhesión celular, polaridad celular, arquitectura tisular y organogénesis [23,24]. En el adulto, Wnt regula la hematopoyesis, osteogénesis, angiogénesis y adipogénesis [25-27]. Este amplio rango de efectos biológicos muestra un alto pleitropismo de las señales de Wnt, las cuales están involucradas en enfermedades humanas incluidas el cáncer. Varios reportes han demostrado la activación aberrante de la vía de señalización de Wnt en diferentes tumores humanos, incluidos el colorectal [28]; gástrico [29] y melanoma [30]. En CaCu se conoce poco acerca del vía de señalización de Wnt, sin embargo, nosotros y otros investigadores hemos descrito recientemente alteraciones de esta vía en neoplasias cervicales. El objetivo de esta revisión, por lo tanto, es analizar la evidencia actual del papel de la vía de Wnt en el desarrollo del CaCU.
Vía de señalización Wnt/beta-catenina.
El receptor de Wnt pertenece a la familia de proteínas Frizzled (FZD), las cuales tienen siete dominios transmembranales. El amplio espectro de procesos celulares regulados por la vía de Wnt puede ser explicado –al menos en parte- por la gran diversidad de ligandos y receptores FZD. En el genoma humano, han sido identificados 19 genes WNT y 11 FZD [31]. Las interacciones entre las proteínas Wnt y sus receptores muestran un importante índice de promiscuidad [32]; por lo tanto, una proteína Wnt puede unirse a diferentes receptores FZD y viceversa. Esta interacción requiere la cooperación de LRP5 y LRP6, proteínas que actúan como correceptores transmembranales [33]. Mutaciones en ambos correceptores pueden conducir al desarrollo de defectos parecidos a las mutaciones que inactivan genes WNT; por ejemplo, defectos en el desarrollo del tálamo dorsal, anormalidades esqueléticas y del tubo neural, disminución en la proliferación de osteoblastos, osteopenia y la vascularización persistente en los ojos del embrión [34-36]. Esta evidencia muestra que las proteínas LRP5/6 juegan un importante papel en la activación y regulación de la vía de Wnt. La interacción entre LRP5/6, FZD y Wnt puede ser regulada negativamente por proteínas secretadas como Dikkopf (Dkk), proteínas secretadas relacionadas a frizzled (sFRP), y Cerberus1 (Cer1), que llegan a inhibir la señalización de esta vía a través de la unión directa a Wnt o a sus correceptores. Dkk se une a LRP junto con otras proteínas transmembranales llamadas Kremens (Krm); de esta manera se promueve la internalización e inactivación de LRP5/6, regulando así esta vía de señalización [37,38].
Actualmente, se conocen tres vías que son activadas después del acoplamiento de Wnt con sus receptores; la determinación de la vía que será activada dependerá de la especificidad del ligando de Wnt con su receptor FZD y muy probablemente de componentes celulares, tales como, coactivadores y correceptores que modulan las señales que llevarán a la activación de determinada vía de señalización.
Estas vías son:
La vía canónica, la cual induce la estabilización y acumulación en el citoplasma de ß-catenina y su posterior translocación al núcleo celular en donde afecta la transcripción de genes blanco (Figura 2) [39].
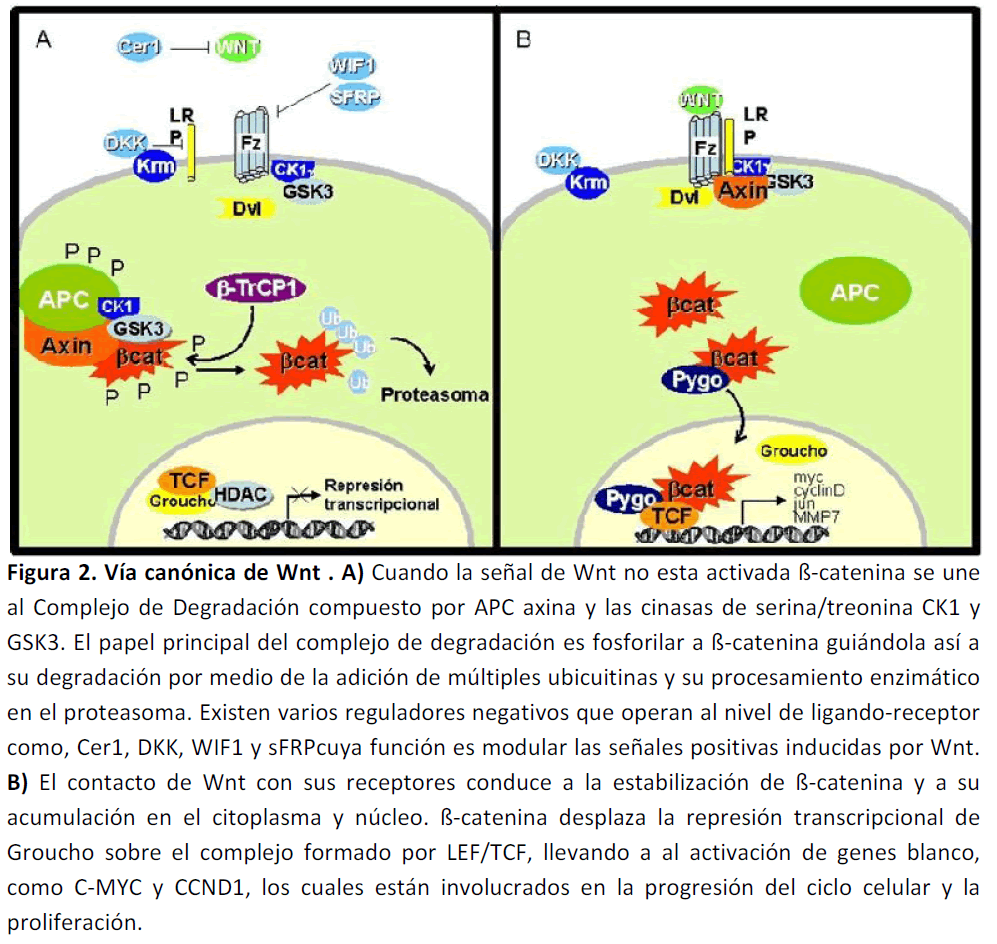
Figura 2. Vía canónica de Wnt . A) Cuando la señal de Wnt no esta activada ß-catenina se une al Complejo de Degradación compuesto por APC axina y las cinasas de serina/treonina CK1 y GSK3. El papel principal del complejo de degradación es fosforilar a ß-catenina guiándola así a su degradación por medio de la adición de múltiples ubicuitinas y su procesamiento enzimático en el proteasoma. Existen varios reguladores negativos que operan al nivel de ligando-receptor como, Cer1, DKK, WIF1 y sFRPcuya función es modular las señales positivas inducidas por Wnt. B) El contacto de Wnt con sus receptores conduce a la estabilización de ß-catenina y a su acumulación en el citoplasma y núcleo. ß-catenina desplaza la represión transcripcional de Groucho sobre el complejo formado por LEF/TCF, llevando a al activación de genes blanco, como C-MYC y CCND1, los cuales están involucrados en la progresión del ciclo celular y la proliferación.
La vía de la polaridad celular planar (PCP), la cual establece la polarización de las células a lo largo del plano de una membrana tisular. Esta vía es importante durante el cierre del tubo neural y la extensión de coclea del oído interno [40].
Finalmente, la vía de Wnt/Ca+2 o “no canónica” la cual regula la adhesión celular y la motilidad mediada por Wnt-5a, dispara la liberación de Ca2+ intracelular para activar enzimas sensibles a Ca2+ como la proteína cinasa C (PKC) y cinasa II dependiente de calmodulina y Ca2+ (CaMKII) sin la activación de la vía de ß-catenina [41]. La vía canónica es la mejor estudiada, además se activa en prácticamente todos los tumores y es el tema central de este trabajo.
Después de la trimerización de Wnt/Fzd/LRPs, el correceptor de LRP es fosforilado guiando a la unión y fosforilación de Disheveled (Dvl), el cual, transduce la señal de Wnt a la célula a través de la unión directa con FZD [42,43]. Dvl es un elemento clave en la amplificación de señales de Wnt a través de efectores específicos [44]. Dvl interactúa con axina, la cual realiza la función de andamiaje molecular por su asociación con proteínas que son clave para la fosforilación y poli-ubicuitinación de ß-catenina, incluyendo GSK-3ß, CK1, APC así como ß-catenina. Cuando no existe señal de Wnt a través de sus receptores, axina se encuentra activa por medio de la fosforilación en residuos específicos, lo que lleva a la inactivación de ß-catenina por la vía del proteasoma. Sin embargo, una vez que se lleva a cabo la trimerización de Wnt/Fzd/LRPs, axina es degradada por la vía del proteasoma; sin embargo, no se conoce aún el mecanismo exacto [45].
Tal vez el evento más importante en el contexto de los procesos moleculares de la activación de la vía de Wnt en tumores, es la estabilización de ß-catenina en el citoplasma y su acumulación en el núcleo. Esto es posible debido a la liberación de ß-catenina del Complejo de Degradación, un conjunto de proteínas ensambladas y activas en ausencia de señales de Wnt, cuya función principal es adicionar ubicuitinas a ß-catenina produciendo su inactivación a través de la vía del proteasoma [24].
En una célula no estimulada, ß-catenina interactúa con E-caderina y ß-catenina en las uniones adherentes, regulando la adhesión celular. El exceso en los niveles de ß-catenina es modulado por medio del Complejo de Degradación [46]. Un componente clave de dicho complejo es APC, que se encuentra unido a axina. Para que el Complejo de Degradación pueda actuar, ß-catenina necesita ser fosforilada por GSK3ß en residuos serina/treonina específicos. Así, ß-catenina fosforilada incrementa dramáticamente su afinidad por APC, desplazando la unión de este con axina pues los motivos de unión de APC a axina y ß-catenina se traslapan [47]. Una gran cantidad de tumores colorrectales contienen mutaciones en este motivo de unión, lo que impide que APC se una a axina o degrade ß-catenina [48].
Después de la activación de FZ/LRP por su ligando, la actividad cinasa del complejo de degradación es inhibida. Consecuentemente, la isoforma no fosforilada de ß-catenina se estabiliza, lo que conlleva al aumento de sus niveles en el citoplasma; esta acumulación conduce a la translocación hacia el núcleo celular de ß-catenina, donde se asocia con el factor de transcripción LEF/TCF y a un activador transcripcional llamado Pygopus (Pygo). Pygo contiene un dominio compartido entre una gran cantidad de proteínas que tienen papel de remodeladores de la cromatina y coactivadores transcripcionales [49]. Mutaciones en Pygo tienen como resultado diversos defectos que son similares a los que se producen por la pérdida de las funciones de Wnt [50].
En la ausencia de la señalización de Wnt, TCF se une a Groucho, formando un complejo represor de los genes que se activan transcripcionalmente por esta vía.
Groucho puede reprimir la transcripción de genes por medio de la inhibición de la maquinaria basal de transcripción y el reclutamiento de desacetilasas de histonas (HDACS) [51,52]. Aunque Groucho no interactúa con el ADN, ejerce su función represiva por su interacción con las regiones amino-terminal de las histonas H3 y 4 y por la modificación de la estructura de la cromatina [53]. De esta manera, la especificidad de los genes que serán reprimidos por Groucho esta dada por LEF/TCF. Cuando ß-catenina entra al núcleo convierte al complejo represor LEF/TCF/Groucho en un complejo activador. Esto es posible debido a que Groucho es desplazado por la interacción de ß-catenina y LEF/TCF, activando así, genes blanco [54]. Varios genes que son activados por la señalización de la vía de Wnt están principalmente involucrados en procesos de proliferación y diferenciación celular; si usted desea consultar una lista bastante completa de genes regulados por esta vía, visite el sitio en línea de Wnt [31].
Vía de Wnt y Cáncer cervical.
Hasta este punto hemos revisado como la vía Wnt/ß-catenina enciende un complejo de señales conduciendo a la activación de genes blanco. Diferentes mutaciones en varios componentes de esta vía han sido estudiadas e identificadas en la mayoría de los tumores humanos, entre ellas mutaciones en APC y CTNNB1. En CaCu solo algunos estudios han mostrado el probable papel que tiene la activación de la vía de Wnt/ß-catenina en su patogénesis. En este aspecto, se ha sugerido que la transformación de queratinocitos humanos inmortalizados por VPH requiere un segundo proceso de activación hacia la malignidad, y esto puede ser llevado a cabo gracias a la activación de la vía canónica de Wnt [55]. Esta hipótesis es apoyada por el hecho que la expresión de ß-catenina se incrementa en 73% de los casos de CaCu, observándose tinción citopásmica y nuclear positiva; no obstante, sólo se encontraron mutaciones en el 20% de los casos analizados [56]. Esto sugiere que la acumulación de ß-catenina puede llevarse a cabo en un nivel superior de la vía de señalización; por ejemplo, inactivación de reguladores negativos como APC o axina. Esta bien establecido que durante el proceso de carcinogénesis distintos genes supresores de tumor son inactivados por metilación anormal de las islas CpG, probablemente debido a un incremento en la actividad de ADN metiltransferasas (DNMT) [57]. E7 de VPH16 tiene la capacidad de unirse e incrementar al actividad de la DNMT1, que es la enzima responsable de adicionar grupos metilo a las islas CpG [58]; de esta manera, resulta factible que reguladores negativos de la vía Wnt/ß-catenina pudieran ser inactivados por un proceso de metilación. En este contexto, los genes sFRPs, axina, DICKKOPF (Dkk) y APC tienen islas CpG enriquecidas en sus promotores, los cuales pueden estar hipermetilados en algunas neoplasias incluyendo CaCu [29,59-61]; por lo tanto, es probable que estos genes puedan ser inactivados por un proceso de metilación en sus promotores, durante la carcinogénesis cervical.
Otro mecanismo involucrado en la activación río arriba de la vía Wnt/ß-catenina es la sobreexpresión de activadores de la vía como los ligandos de Wnt, receptores de frizzled y disheveled. Existe evidencia que muestra la sobreexpresión de WNT10B, -14, FZD10, y DVL-1 en líneas celulares derivadas de CaCu [62-65]; sin embargo, esto aún no ha sido explorado en muestras de tumores por metodologías tradicionales como RT-PCR, inmunohistoquímica o Western y Northern blot.
Recientemente realizamos un análisis de expresión a nivel genómico en CaCu positivo para VPH16 comprarándolo con epitelio cervical normal sin secuencias virales. [66]. En este trabajo pudimos identificar genes y vías celulares con niveles de expresión aberrantes, y una de las vías más alteradas fue Wnt/ß-catenina. Analizamos los niveles de expresión de 55,000 secuencias que representan virtualmente todos los genes expresados en el genoma humano. De esta forma, mostramos un incremento significativo de WNT4, -8a, FZD2, GSK3ß, y ß-catenina en células tumorales. Asimismo, genes que también pertenecen a esta vía son expresados activamente en células cervicales normales como sFRP4, PPP2C, y FZD7 (Figura 3). Esta evidencia demuestras dos importantes hechos: 1) la desregulación en genes específicos que pertenecen a la vía Wnt/ß-catenina podrían jugar un importante papel en la carcinogénesis cervical y 2) la presencia de genes relacionados a Wnt/ß-catenina en tejidos normales sugiere que esta vía esta involucrada en la diferenciación epitelial de las células cervicales. De igual forma, es importante notar que sFRP4, un regulador negativo que inhibe la activación del receptor por medio de la competencia con proteínas de Wnt, es activamente expresado en epitelio normal y ausente en tejidos de CaCu, lo cual indica que este gen pudiera ser importante en el balance de las señales positivas que conducen a la unión de Wnt a su receptor; por consiguiente, regulando la sobre-activación en la señalización que induce a Wnt. Sin embargo, este hecho debe ser comprobado en un número mayor de muestras clínicas de CaCU.
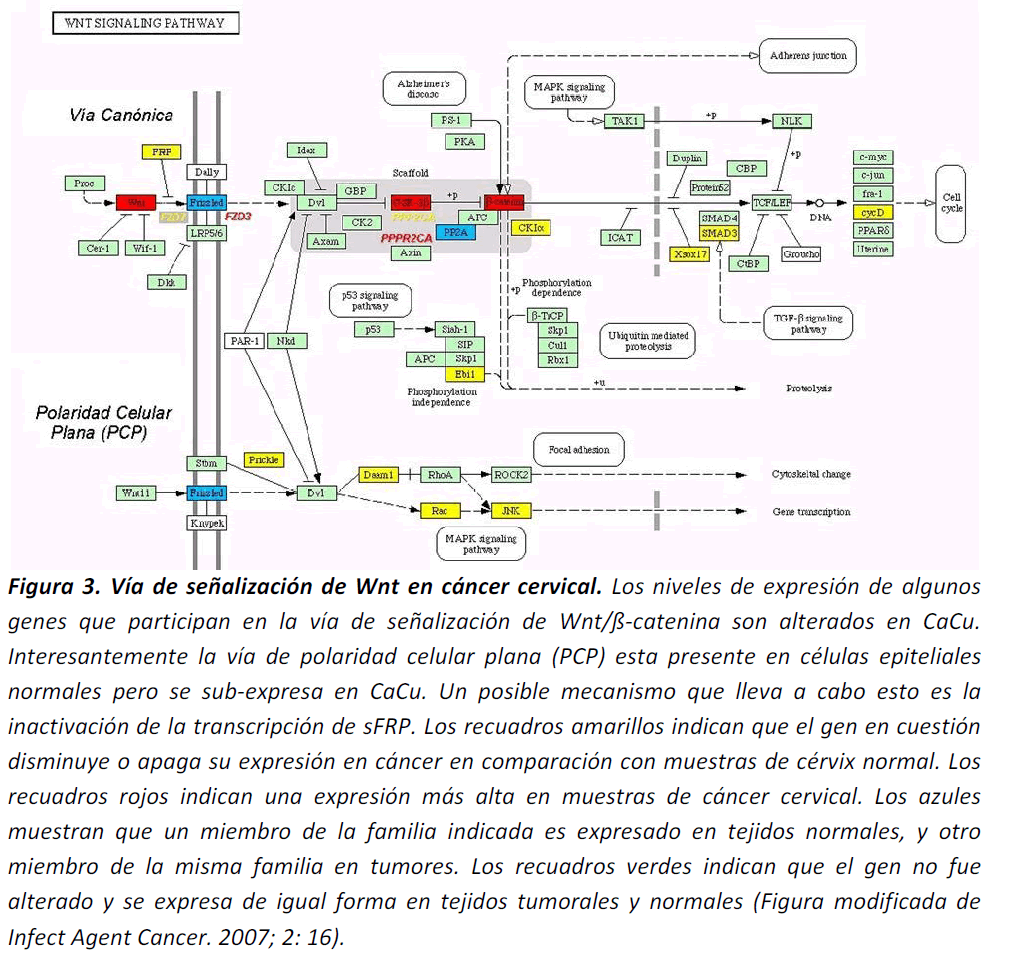
Figura 3. Vía de señalización de Wnt en cáncer cervical. Los niveles de expresión de algunos genes que participan en la vía de señalización de Wnt/ß-catenina son alterados en CaCu. Interesantemente la vía de polaridad celular plana (PCP) esta presente en células epiteliales normales pero se sub-expresa en CaCu. Un posible mecanismo que lleva a cabo esto es la inactivación de la transcripción de sFRP. Los recuadros amarillos indican que el gen en cuestión disminuye o apaga su expresión en cáncer en comparación con muestras de cérvix normal. Los recuadros rojos indican una expresión más alta en muestras de cáncer cervical. Los azules muestran que un miembro de la familia indicada es expresado en tejidos normales, y otro miembro de la misma familia en tumores. Los recuadros verdes indican que el gen no fue alterado y se expresa de igual forma en tejidos tumorales y normales (Figura modificada de Infect Agent Cancer. 2007; 2: 16).
Interesantemente, genes que componen la vía de la polaridad celular plana (PCP) están activamente expresados en tejidos cervicales normales, mostrando que ésta vía de la señalización de Wnt es reprimida en CaCu (Figura 3). En vertebrados PCP se considera como un proceso que afecta la polaridad celular dentro de un epitelio plano e involucra uno o más genes de la vía de PCP. Se ha demostrado que PCP es importante en los procesos de desarrollo y diferenciación de tejidos adultos. Actualmente, los procesos de desarrollo que caen dentro de esos criterios incluyen la extensión convergente el cierre del tubo neural, orientación de las microvellosidades del oído interno y la orientación de los folículos pilosos de la piel [67]. En nuestro conocimiento, no existen reportes previos que muestren genes de PCP activos en epitelio cervical normal. Este resultado demuestra que durante el proceso de carcinogénesis cervical, las células infectadas apagan la vía de PCP, activando la vía canónica con un incremento de los genes que participan en ella, por ejemplo; Wnt4, -8A, FZD2, CTNNB1, entre otros. Este encendido de la vía canónica conduce a la sobre-expresión y activación de genes blanco como MYC, JUN, FOS, y KRAS (Para una lista completa de los genes alterados en CaCu VPH16 por favor consulte el archivo adicional 2 en [66]).
Un mecanismo posible que explique como la vía canónica es privilegiada en CaCu infectado con VPH 16 puede ser la inactivación de sFRP4 en células normales. En un trabajo no publicado, observamos que los inhibidores de sFRP4 y DKK son inactivados por la metilación de su promotor en biopsias de CaCu; cuando los pacientes fueron tratados con inhibidores de DNMTs detectamos una reexpresión de esos genes (datos no publicados). Se ha demostrado que el gen sFRP es frecuentemente inactivado por la metilación de su promotor en cáncer gástrico y hepatocelular (CHC) [29,68]. Asimismo, la restauración de sFRP1 atenúa la señalización de Wnt en células de CHC, disminuye la acumulación anormal de ß-catenina en el núcleo y suprime el crecimiento celular [68]. Como se describió previamente, es común encontrar ß-catenina citoplásmica y nuclear en CaCu acompañada por mutaciones ocasionales en el gen CTNNB1. Con la evidencia previa; podemos especular que la activación de la vía canónica en CaCu se lleva a cabo por medio de la inactivación de reguladores negativos de Wnt; es decir, los genes sFRP y particularmente SFRP4 y DKK, como lo hemos mostrado [66,69].
Conclusiones
Aunque el uso extenso de exámenes de Papanicolau y colposcópicos han disminuido significativamente los índices de mortalidad, el CaCu sigue siendo la segunda causa de muerte de mujeres a nivel mundial. Se han encontrado secuencias de VPH en el 99% de las muestras de CaCu y la infección con VPH es el factor etiológico más importante en la carcinogénesis cervical. Asimismo, la infección con VPH es muy común entre la población sexualmente activa, pero solo una pequeña fracción de individuos infectados desarrollara CaCu. De esta forma, VPH puede ser considerado como el evento inicial en el proceso de carcinogénesis secuencial que conduce al desarrollo de CaCu. Las vías moleculares involucradas en la progresión de células infectadas con VPH a carcinoma invasor aún no han sido claramente identificadas. En esta revisión, analizamos el papel de la activación de la vía de Wnt/ß-catenina y la inactivación de PCP en células de carcinoma invasor como un segundo proceso molecular requerido para el desarrollo del CaCu.
En esta neoplasia, las mutaciones en el gen CTNNB1 son poco frecuentes; por lo que la activación de la vía de Wnt parece llevarse a cabo en los componentes río arriba de ß-catenina. Este hecho podría ser realizado por la inactivación de reguladores negativos de la vía o por la sobrexpresión de elementos que activan a la misma.
Otra rama que puede ser relevante en la señalización de la vía de Wnt durante la patogénesis del CaCu es la vía PCP, la cual está involucrada en procesos de diferenciación. PCP es un proceso de diferenciación y morfogénesis clave en el desarrollo epitelial. En epitelio cervical normal, las células se encuentran polarizadas y migran del espacio basal hacia el luminal conforme se diferencian. Interesantemente, los genes componentes de la vía de PCP están subexpresados en CaCu, indicando que este mecanismo puede ser abatido antes del establecimiento del tumor. Desde el punto de vista del diagnóstico este hecho puede ser importante, debido a que si es posible observar este hecho como un evento temprano, podríamos contar con marcadores moleculares potenciales para la detección temprana.
Contribución de los autores.Contribución de los autores.
CPP concibió y escribió el manuscrito; ADG y JBM participaron en la discusión y análisis del contenido.
Agradecimientos.
Agradecemos a Itzel Pérez-Rodríguez por su ayuda en el diseño de las figuras de este trabajo. Durante este trabajo CPP fue destinatario del programa posdoctoral de la Universidad Nacional Autónoma de México.
1080
References
- Parkin D, Bray F: Chapter 2: the burden of HPV-related cancers. Vaccine 2006, 24:S11-S25.
- Parkin D, Bray F, Ferlay J, Pisani P: Estimating the world cancer burden: Globocan 2000. Int J Cancer 2001, 94:153-156.
- Kessler I: Venereal factors in human cervical cancer: evidence from marital clusters. Cancer 1977, 39:1912-1919.
- zur Hausen H, Meinhof W, Scheiber W, Bornkamm G: Attempts to detect virus-specific DNA sequences in human tumors: I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer 1974, 13:650-656.
- zur Hausen H: Condylomata acuminata and human genital cancer. Cancer Res 1974, 36:794.
- Bosch FX, Muñoz N, Shah K, Meheus A: Second International Workshop on the epidemiology of cervical cancer and human papillomavirus. Int J Cancer 1992,171-173.
- Walboomers J, Jacobs M, Manos M, Bosch FX, Kummer J, Shah K, et al.: Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999,12-19.
- Bernard H, Chan S, Manos M, Ong C, Villa L, Delius H, et al.: Identification and assessment of known and novel human papillomaviruses by polymerase chain reaction amplification, restriction fragment length polymorphisms, nucleotide sequence, and phylogenetic algorithms. J Infect Dis 1994, 170:1077-1085.
- Munoz M, Bosch F, de San Jose S, Vergara A, del Moral A, Munoz M, et al.: Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003,518-527.
- Molano M, Van den Brule A, Plummer M, Weiderpass E, Posso H, Arslan A, et al.: HPV Study Group. Determinants of clearance of human papillomavirus infections in Colombian women with normal cytology: a population-based, 5-year follow-up study. Am J Epidemiol 2003, 158:486-494.
- Ho G, Bierman R, Beardsley L, Chang C, Burk R: Natural history of cervicovaginal papillomavirus infectionin young women. N Engl J Med 1998, 338:423-428.
- Matsukura T, Koi S, Sugase M: Both episomal and integrated forms of Human Papillomavirus Type 16 are involved in Invasive Cervical cancers. Virology 1989, 172:63-72.
- Ward P, Coleman DV, Malcolm DB: Regulatory mechanisms of the papillomaviruses. Trends Genet, 1989, 5:97-98.
- Stevenson M, Hudson L, Burns J, Stewart R, Wells M, Maitland N: Inverse relationship between the expression of the human papillomavirus type 16 transcription factor E2 and virus ADN copy number during the progression of cervical intraepithelial neoplasia. J Gen Virol 2000, 81:1825-1832.
- zur Hausen H: Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst 2000, 92:690-698.
- Park T, Fujiwara H, Wright T: Molecular biology of cervical cancer and its precursors. Cancer 1995, 76:1902-1913.
- Hudelist G, Manavi M, Pischinger K, Watkins-Riedel T, Singer C, Kubista E, et al.: Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol Oncol 2008, 92:873-880.
- Ueda Y, Enomoto T, Miyatake T, Ozaki K, Yoshizaki T, Kanao H, et al.: Monoclonal expansion with integration of high-risk type human papillomavirus is an essential step for cervical carcinogenesis: association of clonal status and human papillomavirus infection with clinical outcome in cervical intraepithelial neoplasia. Lab Invest 2003, 83:1517-1527.
- Wentzensen N, Vinokurova S, von Knebel Doeberitz M: Systematic Review of Genomic Integration Sites of Human Papillomavirus Genomes in Epithelial Dysplasia and Invasive Cancer of the Female Lower Genital Tract. Cancer Res 2004, 64:3878-3884.
- Scheffner M, Werness B, Huibregtse J, Levine A, Howley P: The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63:1129-1136.
- Dyson N, Howley PM, Munger K, Harlow E: The human papilloma virus-l6 El oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243:934-940.
- Nusse R, van Ooyen A, Cox D, Fung Y, Varmus H: Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature 1984, 307:131-136.
- Wodarz A, Nusse R: Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol 1998, 14:59-88.
- Peifer M, Polakis P: Wnt signaling in oncogenesis and embryogenesis: a look outside the nucleus. Science 2000, 287:1606-1609.
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al.: Inhibition of Adipogenesis by Wnt Signaling. Science 2000, 289:950-953.
- Goodwin A, D'Amore P: Wnt signaling in the vasculature. Angiogenesis 2002, 5:1-9.
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al.: LDL Receptor-Related Protein 5 (LRP5) Affects Bone Accrual and Eye Development. Cell 2001, 107:513-523.
- Segditsas S, Tomlinson I: Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25:7531-7537.
- Nojima M, Suzuki H, Toyota M, Watanabe Y, Maruyama R, Sasaki S, et al.: Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007, 26:4699-4713.
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P: Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275:1790-1792.
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, et al.: A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382:225-230.
- Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, et al.: Arrow encodes an LDL-receptor-related protein essential for Wingless signaling. Nature 2000, 407:527-530.
- Zhou C, Pinson K, Pleasure S: Severe defects in dorsal thalamic development in low-density lipoprotein receptor-related protein-6 mutants. J Neurosci 2004, 24:7632-7639.
- Kato M, Patel M, Levasseur R, Lobov I, Chang B, Glass D, et al.: Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol 2002, 157:303-314.
- Kokubu C, Heinzmann U, Kokubu T, Sakai N, Kubota T, Kawai M, et al.: Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development 2004, 131:5469-5480.
- Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, et al.: Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signaling. Nature 2002, 417:664-667.
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC: An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407:535-538.
- Qian D, Jones C, Rzadzinska A, Mark S, Zhang X, Steel K, et al.: Wnt5a functions in planar cell polarity regulation in mice. Dev Biol 2007, 306:121-133.
- Kuhl M, Sheldahl L, Park M, Miller J, Moon R: The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape. Trends Genet 2000, 16:279-283.
- Umbhauer M, Djiane A, Goisset C, Penzo-Mendez A, Riou J, et al: The C- terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/ betacatenin signalling. EMBO J 2000, 19:4944-4954.
- Wallingford JB, Habas R: The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development 2005, 132:4421-4436.
- Wharton KA: Runnin' with the Dvl: pr oteins that associate with Dsh/Dvl and their significance to Wnt signal tr ansduction. Dev Biol 2003, 253:1-17.
- Yamamoto A, Nagano T, Takehara S, Hibi M, Aizawa S: Shisa promotes head formation through the inhibition of receptor protein maturation for the caudalizing factors, Wnt and FGF. Cell 2005, 120:223-235.
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R: beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 1997, 16:3797-3804.
- Xing Y, Clements WK, Kimelman D, Xu W: Crystal structure of a [beta]-catenin//Axin complex suggests a mechanism for the [beta]-catenin Destruction complex. Genes Dev 2003, 17:2753-2764.
- Kinzler KW, Vogelstein B: Lessons from Hereditary Colorectal Cancer. Cell 1996, 87:159-170.
- Belenkaya TY, Han C, Standley HJ, Lin X, Houston DW, Heasman J, et al.: pygopus encodes a nuclear protein essential for Wingless/Wnt signaling. Development 2002, 129:4089-4101.
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, et al.: Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 2002, 109:47-60.
- Yu X, Li P, Roeder R, Wang Z: Inhibition of androgen receptor-mediated transcription by amino-terminal enhancer of split. Mol Cell Biol 2001, 21:4614-4625.
- Chen G, Fernandez J, Mische S, Courey A: A functional interaction between the histone deacetylase Rpd3 and the corepressor groucho in Drosophila development. Genes Dev 1999, 13:2218-2230.
- Sekiya T, Zaret KS: Repression by Groucho/TLE/Grg proteins: genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol Cell 2007, 28:291-303 .
- Daniels DL, Weis WI: Neta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol 2005, 12:364-371.
- Uren A, Fallen S, Yuan H, Usubutun A, Kucukali T, Schlegel R, et al.: Activation of the Canonical Wnt Pathway during Genital Keratinocyte Transformation: A Model for Cervical Cancer Progression. Cancer Res 2005, 65:6199-6206.
- Shinohara A, Yokoyama Y, Wan X, Takahashi Y, Mori Y, Takami T, et al.: Cytoplasmic/nuclear expression without mutation of exon 3 of the beta-catenin gene is frequent in the development of the neoplasm of the uterine cervix. Gynecol Oncol 2001,-450.
- Robertson K: DNA methylation, methyltransferases, and cancer. Oncogene 2001,20-3139.
- Burgers W, Blanchon L, Pradhan S, Launoit Y, Kouzarides T, Fuks F: Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26:1650-1655.
- Oates NA, van Vliet J, Duffy DL, Kroes HY, Martin NG, Boomsma DI, et al.: Increased DNA Methylation at the AXIN1 Gene in a Monozygotic Twin from a Pair Discordant for a Caudal Duplication Anomaly. The American Journal of Human Genetics 2006, 79:155-162.
- Yang HJ, Liu V, Wang Y, Tsang P, Ngan H: Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer 2006, 6:212.
- Lee J, Yoon YS, Chung JH: Epigenetic silencing of the WNT antagonist DICKKOPF-1 in cervical cancer cell lines. Gynecologic Oncology 2008, 109:270-274.
- Kirikoshi H, Katoh M: Expression and regulation of WNT10B in human cancer: up-regulation of WNT10B in MCF-7 cells by beta-estradiol and down-regulation of WNT10B in NT2 cells by retinoic acid. Int J Mol Med 2002, 10:507-511.
- Kirikoshi H, Sekihara H, Katoh M: Expression of WNT14 and WNT14B mRNAs in human cancer, up-regulation of WNT14 by IFNgamma and up-regulation of WNT14B by beta-estradiol. Int J Oncol 2001, 19:1221-1225.
- Koike J, Takagi A, Miwa T, Hirai M, Terada M, Katoh M: Molecular cloning of Frizzled-10, a novel member of the Frizzled gene family. Biochem Biophys Res Commun 1999, 262:39-43.
- Okino K, Nagai H, Hatta M, Nagahata T, Yoneyama K, Ohta Y, et al.: Up-regulation and overproduction of DVL-1, the human counterpart of the Drosophila dishevelled gene, in cervical squamous cell carcinoma. Oncol Rep 2003, 10:1223.
- Perez-Plasencia C, Vazquez-Ortiz G, Lopez-Romero R, Pina-Sanchez P, Moreno J, Salcedo M: Genome wide expression analysis in HPV16 Cervical Cancer: identification of altered metabolic pathways. Infectious Agents and Cancer 2007, 2:16.
- Wang Y, Nathans J: Tissue/planar cell polarity in vertebrates: new insights and new questions. Development 2007, 134:647-658.
- Shih Y, Hsieh C, Lai H, Yan M, Hsieh T, Chao Y, et al.: SFRP1 suppressed hepatoma cells growth through Wnt canonical signaling pathway. Int J Cancer 2007, 121:1028-1035.
- Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, de la Cruz-Hernandez E, Herrera L: Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev 2008, 34:206-222.

