Key words
|
| |
| Biomarker, Drug Discovery and Development, New Molecular Entity, Alphalisa Technology, High Throughput Screening. |
| |
Introduction
|
| |
| Biological markers (biomarkers) have been defined by Hulka and colleagues1 as “cellular, biochemical or molecular alterations that are measurable in biological media such as human tissues, cells, or fluids.” More recently, the definition has been broadened to include biological characteristics that can be objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention.2 In practice, biomarkers include tools and technologies that can aid in understanding the prediction, cause, diagnosis, progression, regression, or outcome of treatment of disease. For the nervous system there is a wide range of techniques used to gain information about the brain in both the healthy and diseased state. These may involve measurements directly on biological media (e.g., blood or cerebrospinal fluid) or measurements such as brain imaging which do not involve direct sampling of biological media but measure changes in the composition or function of the nervous system.3 |
| |
| Biomarkers of all types have been used by generations of epidemiologists, physicians, and scientists to study human disease. The application of biomarkers in the diagnosis and management of cardiovascular disease, infections, immunological and genetic disorders, and cancer are well known.1,3 Their use in research has grown out of the need to have a more direct measurement of exposures in the causal pathway of disease that is free from recall bias, and that can also have the potential of providing information on the absorption and metabolism of the exposures.4 Neuroscientists have also relied on biomarkers to assist in the diagnosis and treatment of nervous system disorders and to investigate their cause. Blood, brain, cerebrospinal fluid, muscle, nerve, skin, and urine have been employed to gain information about the nervous system in both the healthy and diseased state. This paper focuses on biomarkers as defined by Hulka et al.,1 i.e., direct measures of biological media, and other papers in this issue will address brain imaging and other markers. The rapid growth of molecular biology and laboratory technology has expanded to the point at which the application of technically advanced biomarkers will soon become even more feasible.5–8 Molecular biomarkers will, in the hands of clinical investigators, provide a dynamic and powerful approach to understanding the spectrum of neurological disease with obvious applications in analytic epidemiology, clinical trials and disease prevention, diagnosis, and disease management. |
| |
|
⇒ What exactly are biomarkers?
|
| |
| ⇒ Gene expression products (mRNAs) |
| |
| ⇒ Genetic defects (mutations, SNPs, chromosomal anomalies) |
| |
| ⇒ Metabolites (drugs; endogenous) |
| |
| ⇒ Micro-RNAs |
| |
| ⇒ Proteins and their isoforms |
| |
| ⇒ Altered cellular distributions |
| |
| What is a “Clinical Endpoint”? |
| |
| “A Characteristic or variable that reflects patient feeling, function or survival” |
| |
| Biomarkers can be “Surrogate endpoints”: |
| |
| · Biomarker intended to substitute for a clinical endpoint (predict benefit or harm) based on epidemiologic, therapeutic, pathophysiologic or other scientific evidence |
| |
| · Biomarker Examples: Cholesterol, Blood pressure levels for heart disease, PSA (antigen) for prostate cancer, HbA1c in diabetes, CD4 lymphocyte count for AIDS |
| |
| · How are they used in drug development? |
| |
| Once a proposed biomarker has been validated, it can be used to diagnose disease risk, presence of disease in an individual, or to tailor treatments for the disease in an individual if a treatment alters the biomarker, which has a direct connection to improved health, the biomarker serves as a "surrogate endpoint" for evaluating clinical benefit.9 |
| |
|
ncorporation of Biomarkers in Drug Development
|
| |
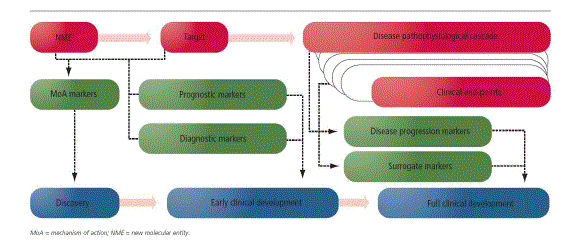
| Over the past decade or so, biomarkers have gained increased visibility and importance in pharmaceutical drug development across therapeutic areas, spanning all stages of development. Aided by recent advances in numerous fields, including pharmacogenomics, imaging and molecular diagnostics, as well as advances in the understanding of the pathophysiology of diseases at the molecular level, biomarkers can enhance the value of all phases of a drug development programme in a variety of ways by enabling validation of mechanism of action (MoA), identification of prognostic signatures, patient selection/stratification to enhance clinical response, development of diagnostic assays, monitoring of disease progression and response to therapy and development of surrogate biomarkers. In addition, biomarkers have the potential to have a significant impact on the commercial value of a programme by enhancing product differentiation, market position and payer reimbursement. As a result, biomarkers are now routinely included in research and development (R&D) strategies for new molecular entities (NMEs), from drug discovery to regulatory approval and beyond (Figure-1). However, for a project manager supporting a drug development project, biomarkers can add significant complexity to project planning, scenario analysis, risk assessment/management and go/no go recommendations.10 |
| |
TYPES OF BIOMARKERS:-
|
| |
|
Prognostic and Epidemiological Biomarkers
|
| |
| These biomarkers are key to providing information on the outcome of a disease irrespective of therapy. As an NME progresses from discovery towards development, a key part of strategy development involves the selection of target disease and indications. Based on the understanding of the MoA of the NME, usually a disease with unmet medical need is targeted. In recent advances diseases can be understand at the cellular and molecular levels and here prognostic and epidemiological biomarkers should be factored in to R & D strategy development. These biomarkers can play a key role in defining the right segment of the patient population whose unmet need could be met by a given NME with a specific MoA. As a result, strategy development for an NME needs to take into account prognostic and epidemiological biomarkers.11 |
| |
|
Diagnostic Biomarkers
|
| |
| Diagnostic biomarker have key role in a suitable target patient population has been identified for an NME, the next question is how best to identify those patients who might derive therapeutic benefits from the specific NME. Diagnostic biomarkers cover a wide range of markers, from those that could be measured via routine tests. When a development of diagnostic kit as a companion product, a coordinated development strategy needs to be developed as early as possible in the development cycle, with ongoing, real time communication and collaboration between R&D personnel involved in both arms of the development effort. One of the most well-known examples of a diagnostic biomarker playing a crucial role in the development and approval of an NME involves the assessment of human epidermal growth factor receptor-2 (HER-2) for treatment of breast cancer in HER-2- positive patients with trastuzumab. Since it is directed against HER-2, assessment of the HER-2 status of the patient is essential to rule trastuzumab in or out as a potential treatment option.12-17 |
| |
|
Mechanism of Action Markers
|
| |
| When an NME is selected for development, the R&D project team usually has access to literature-based as well as internally generated pre-clinical data based on which a hypothesis for the MoA of the NME is developed. Markers based on the MoA are also often closely linked to pharmacodymanic markers that allow the establishment of a chain of molecular and biochemical events, also known in literature as the pharmacological audit trail, starting from the entry of the NME into the blood stream, to the target tissue, to the impact on cellular and molecular targets and, subsequently, to the impact on the pathophysiological cascade of the target disease. Thus, these markers are also known as proof-ofprinciple markers as they help to establish the principle for the potential activity of an NME via an intended pathway. MoA markers are also categorized as proximal or target engagement markers, given their role in testing the hypothesis on the interaction of the NME with the molecular target. This hypothesized MoA is tested in animal models and subsequently in early clinical trials. |
| |
| These studies are expansive, and also take a significant time, but a properly designed study can be enormously beneficial in making decision to accelerate, slow down and terminate the development of NME.18 |
| |
|
Disease Progression Markers and Response to Therapy Markers
|
| |
| In addition to the MoA markers, pursuing the pharmacological audit trail leads to a second set of markers that are associated with the pathophysiology of the target disease. These markers are also referred to as distal biomarkers given their relatively late involvement in the pathophysiological cascade. In the case of MoA markers the effect of NME or it’s interactions with the target, when in diseases progression markers are developed to monitor the disease at molecular, cellular, and target tissue levels. Activity based on the response to therapy markers can enable a team to potentially initiate larger trials or trigger advanced stages of the same trial if an adaptive design is employed. On the other hand, lack of activity based on these biomarkers could also allow an early termination of development.19-21 |
| |
|
Surrogate Biomarkers
|
| |
| Surrogate markers are similar to the response to therapy biomarkers except that these markers are specifically pursued as primary endpoints and potential surrogates for meaningful clinical benefit in a given indication and are often used to seek regulatory approval for an investigational agent. In certain cases, it is possible to validate an exploratory biomarker as a surrogate biomarker by demonstrating correlation between biomarker-based activity and approvable clinical end-points.22 |
| |
|
Impact of Biomarkers on Drug Development
|
| |
| This approach requires taking a comprehensive look at the development cycle as a whole, rather than the fragmented approach of assessing the merits and designs of pre-clinical and clinical studies one at a time. This approach requires building an overall strategy that combines discovery, toxicology, pharmacology, diagnostic, clinical and regulatory perspectives throughout the R&D process so that each individual study is not only consistent with but also complementary or supplementary to the rest of the studies. |
| |
| Incorporation of biomarkers into a drug discovery and development programme has an impact on these three major tenets of project management. Testing and analysis of samples (genomic, proteomic, pharmacokinetic, tissue staining, imaging, etc.) to support biomarkers are often costly and resourceintensive. Biomarkers have the capacity to significantly alter the scope of a programme due to their potential impact on patient selection or stratification, go/no go decisions and even regulatory approvals. Finally, biomarkers have a significant impact on timelines, not only due to additional work on their identification, development and potential validation, but also due to their potential to accelerate the development process through early decisions to progress to the next phase of development and use of surrogate biomarkers to gain accelerated regulatory approval.16,23 |
| |
|
Incorporating biomarkers into drug discovery and development
|
| |
| The ultimate biomarker would be able to indicate the diseased state and be altered by therapeutic intervention so that clinical outcome could be predicted. However, it is most unlikely that one biomarker could be used for all these purposes; different biomarkers must be used at different stages in drug discovery and development. Biomarkers have been classified into three types. The first are markers of the disease state. The second type indicates the effects of a therapeutic intervention based on the mechanism of action for a drug, even though this may not be known to be associated with the desired clinical outcome. This type of biomarker is often used in preclinical screening of drug candidates. The third type of biomarker is used as a surrogate end-point because a change in that marker predicts clinical outcome. Areas like oncology have progressed further than other areas in biomarker discovery and use, and have already proven successful with such examples as HER-2, a biomarker for a subset of more aggressive breast cancers. HER-2, a human epidermal growth factor, not only identifies patients that will benefit from Herceptin but is also the target of the drug. Not all diseases will be amenable to biomarker discovery and use. Biomarkers will be easier to use in diseases that are well defined with more homogenous patient populations.17 |
| |
|
Preclinical stage
|
| |
| Incorporating biomarkers early in the discovery process when a new therapeutic target is being identified is of utmost importance especially if the biomarkers can reflect mechanism-based intervention. To find these discovery biomarkers, researchers can look to their pre-clinical experiments, such as animal and cell culture models. Analysis of expression changes in target versus nontarget tissues in treated animal models may provide possible information to establish biomarkers for a potential therapeutic. High-throughput screening targets may also be used as an initial source of potential discovery biomarkers. Here discovery biomarkers which are used preclinically may not be the same biomarkers used after the drug is selected to enter clinical trials. In preclinical studies, biomarkers are used to determine if the drug is hitting the target, after which additional biomarkers must confirm that hitting the target actually alters the pathophysiological mechanism, and that altering this mechanism affects clinical outcome.18 |
| |
|
Clinical stage
|
| |
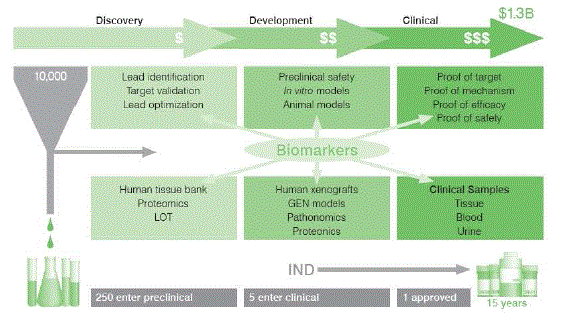
| For one successful drug, there are 60 in discovery, 20-30 in early development, and 5-8 in clinical programmes. One of the biomarker development goals is to assess drug safety and efficacy accurately, thereby reducing attrition of drugs during clinical phases of development and hence reducing the overall cost of drug development. Singulex’ new, more sensitive technologies developed for protein and metabolite biomarker detection provides a significant benefit for monitoring established safety and toxicity biomarkers, such as troponin I for cardio toxicity, throughout clinical trials. The current cardio toxicity tests can only detect large-scale damage, which halts drug development. The benefits of more sensitive assays are not only the detection of mild toxicity, repeated exposure to which could lead to major problems, but also in giving the green light for further development. Being able to detect normal levels of biomarkers, especially for toxicity, but for any deviation from normal provides the total information needed to make accurate decisions on whether to proceed further in drug development. Another goal, as the drug and biomarkers move through the drug development phase and into clinical trials, is to identify and stratify patients to maximize the signal in early proof of- concept trials. If biomarkers could identify patients whose response rate could be double or higher, then clinical trials could involve half the current number of subjects yet yield sufficient proof of efficacy.19 |
| |
|
Companion diagnostics
|
| |
| The use of biomarkers to identify and stratify patients in clinical trials would ultimately create drug/diagnostic combinations, such as Herceptin/ HER-2, and fully deliver the promises of personalized medicine. Today’s drugs which have been withdrawn from market or Phase 3 development due to serious adverse events may have had a different fate if combined with a companion diagnostic to identify responders or to closely monitor toxicity.20 |
| |
|
Partnering strategies
|
| |
| The development and use of biomarkers in drug discovery and development has fallen into a gray area where no one knows who is responsible for developing the technology and assays within a company. For effective biomarker integration into drug discovery and development, pharmaceutical companies are seeking collaboration, not only within their company but also with new companies and technologies. Singulex provides the technology to validate and deliver highly sensitive, customizable asssays for almost any protein or metabolite biomarker. With numerous collaborations in leading pharma and academic institutions, assays for cardiac and liver toxicity have been developed as well as for biomarkers for diseases such as pancreatic cancer and Alzheimer’s disease.21 |
| |
|
The Role of Biomarkers in Drug Discovery and Development
|
| |
| Biomarkers have been used in drug development and treatment of diseases for a long time, the identification of new predictive safety and efficacy biomarkers is expected to reduce the time and cost of drug development. In addition, the use of novel, but less well-established, pharmacodymanic biomarkers can further facilitate decision-making from discovery through preclinical development and in to clinical trials, while rapid advance in genomic and proteomic have increased the discovery of new biomarkers and their value in drug development and treatment of diseases. Biomarker measurements now support target validation and proof of target, mechanism, and efficacy, and they are being developed first in preclinical animal model of diseases. The majority of biomarker research is done in clinical trials test cancer drugs, which represents the single largest therapeutic class of drugs in development.22 |
| |
| The drug discovery funnel continues to be in undated with novel compounds (Figure-2). High-throughput screening (HTS) is under escalating pressure to screen more targets against more compounds. It is reported that out of 35 million compounds screened, 5000 hits are identified. Of this 5000 hits, only 5 become drug candidate that make it to human testing. Only one of these 5 is approved for human use. On the average, it takes 10-15 years at an estimated cost of $1.3 billion to get one new drug from molecules to medicine. A primary goal with in Charles River is to help our customers move their products in to clinical trials faster and with greater probability for success.23 |
| |
| The drug discovery funnel continues to be in undated with novel compounds (Figure-2). High-throughput screening (HTS) is under escalating pressure to screen more targets against more compounds. It is reported that out of 35 million compounds screened, 5000 hits are identified. Of this 5000 hits, only 5 become drug candidate that make it to human testing. Only one of these 5 is approved for human use. On the average, it takes 10-15 years at an estimated cost of $1.3 billion to get one new drug from molecules to medicine. A primary goal with in Charles River is to help our customers move their products in to clinical trials faster and with greater probability for success.23 |
| |
|
AlphaLISA™ in Biomarker Detection for Drug Discovery
|
| |
| Enzyme-linked immunosorbent assays (ELISAs) have historically been among the most effective and widely adopted assays for use in detection and quantification of low analyte concentrations. The technology is selective, sensitive and versatile; however, its usefulness has been limited by low throughput due to wash steps, a generally narrow dynamic range and the inability to use low-affinity antibodies. |
| |
| Due to these limitations, biomarker detection in drug discovery has necessarily employed more time- and resource-intensive processes. In general, a homogeneous (no wash) technology that could detect a wide range of biological analytes would be of considerable value to drug discovery programmes.24 |
| |
| To utilize the advantages of ELISA assays, including their inherent simplicity and economy, yet reduce the disadvantages, PerkinElmer, Inc. (PKI) developed AlphaLISA™. The technology does not require wash steps and is easy to miniaturize and automate, enabling an efficient high throughput screening (HTS) set-up. AlphaLISA can be set up as either sandwich or Competition immunoassays and be used to detect analytes without the removal of biological matrices such as serum, plasma or cell lysates.25 |
| |
|
AlphaLISA for Biomarker Analysis
|
| |
| AlphaLISA is specifically designed to meet the requirements for high throughput assays for detecting analytes of various sizes, from a small molecule such as oestrogen, to protein–protein complexes, to full-size phages. Furthermore, AlphaLISA is ideal for complex samples such as serum and plasma. |
| |
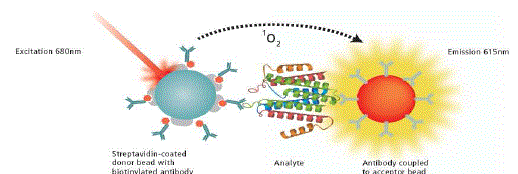
| The technology is based on PKI’s Amplified Luminescent Proximity Homogenous Assay (Alpha Screen®) and employs oxygen-channeling chemistry developed initially as a diagnostic detection assay platform under the name of LOCI®. 1–2 AlphaLISA exploits the short diffusion distance of singlet oxygen to initiate a chemiluminescent reaction near the site where it was formed. The technology comprises two discrete polystyrene beads, designated as ‘Donor’ and ‘Acceptor’. Each Donor/Acceptor pair can be separated by as much as 200nm and retain efficient energy transfer. This relatively large distance allows greater flexibility in the choice of analyte that can be studied, and thus accommodates assays for larger molecules such as full-length proteins; immunocomplexes and others (Figure-1). |
| |
| As the lifetime of the singlet oxygen reactive species in water is short (approximately four minutes), the Donor and Acceptor beads need to be bound to one another to generate a signal. Beads that do not bind exhibit a very low singlet oxygen concentration, which contributes minimally to the background signal. AlphaLISA emission is intense and better defined spectrally (615nm) than traditional ELISA technology, and is less prone to matrix interferences from compounds such as haemoglobin or transferrin. AlphaLISA assays require only small sample volumes (5μl), yet their analytical range is more than 100-fold greater than that of ELISA. As AlphaLISA is highly amplified and can employ the same antibody pairs, assays using this approach are as selective as conventional ELISAs, but more sensitive.26 |
| |
|
AlphaLISA and Cellular Kinase Assays
|
| |
| AlphaLISA has shown excellent results cell signaling events in complex samples such as phosphorylation– as well as proteolysis, ubiquitination, sumoylation and glycosylation thus opens pathway mapping to new levels of simplicity, economy and precision. Several technologies besides AlphaLISA have also been developed as alternatives to ELISA. |
| |
| AlphaScreen bead-based assays (PerkinElmer) were developed to detect the function of both kinase families.5–8 AlphaScreen can be separated by as much as 200nm and retain efficient energy transfer. AlphaLISA wavelength range is 520-620 nm. |
| |
| Alphalisa has two other technologies of note are the homogenous fluorometric micro volume assay technology (FMAT) and enhanced electrochemiluminescence (ECL).9,10 AlphaLISA, FMAT and ECL technologies are highly sensitive, do not require extensive washing and can be formatted for HTS. Beasley et al. presented recent preliminary data that compared the ability of FMAT, ECL and AlphaLISA methods to detect common biomarkers.11 Here, AlphaLISA exhibited higher sensitivity and required lower sample volumes than the other assays. They also indicated that ECL and FMAT techniques involved the use of specialized detection instrumentation (laser scanning microscopy for FMAT) and equipment (electrochemical plates for ECL), which limited their flexibility and increased assay cost. ECL was also limited by the high costs of the consumable electrochemical plates.24,27 |
| |
|
Merits Of AlphaLISA
|
| |
| The main concern of AlphaLISA is that it is sensitive to intense light or long exposure to ambient light, a problem that is easily overcome by simple assay adjustments. Singlet oxygen can be sequestered by compounds in screening libraries that can scavenge radical oxygen. Donor bead photo bleaching can be a limitation as system is effectively limited to a single read. Nonetheless, AlphaLISA has greater flexibility than technologies such as ECL and FMAT, all three of which require a high-energy laser excitation source.27-30 |
| |
| The greatest advantage of AlphaLISA is that it is applicable to a broad range of analytes.31-35 The assays are homogeneous (no wash), rapid, highly robust and more sensitive than previously reported immunoassay methods. They are economical from both a reagent use and assay time perspective, and are ideal for HTS applications. Furthermore, AlphaLISA assays do not require insertion of large fluorescent epitope tags that can sterically hinder the molecular interactions.36 AlphaLISA can be employed in crude biological fluids such as cell lysates, serum and plasma to measure enzyme activity and cellular/body fluid matrices, which do not easily affect the assay readout.37-40 |
| |
Conclusion
|
| |
| AlphaLISA can be used to measure a diverse range of molecular interactions of interest across drug discovery. The homogenous nature of the technique allows it to be an important tool in HTS of new small molecules and, most recently, of novel protein therapeutics. For example, it has been used for hybridoma screening for thousands of clones that express antibodies for therapeutic development.41-45 Such screening currently involves use of conventional ELISAs, which, as noted above, are less adaptable for high-capacity screening and potentially more costly. Cell signaling events in complex samples such as phosphorylation, proteolysis, ubiquitination, sumoylation and glycosylation remain difficult to measure which can be also covered by AlphaLISA.46-48 |
| |
Conflict of Interest
|
| |
| NIL |
| |
Source of Support
|
| |
| NONE |
| |
Tables at a glance
|
 |
| Table 1 |
|
| |
Figures at a glance
|
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
|
| |


