Keywords
Angioma; Retina; Von Hippel-Lindau disease
Introducción
Los angiomas de retina son lesiones vasculares benignas que se presentan como tumoraciones vasculares redondas, bien limitadas, localizadas en la periferia de la retina o encima de la papila fihura [1], solitarias, de crecimiento lento y con el tiempo pueden causar disminución de la visión por efecto de tracción o exudativo en la retina [2], el tratamiento consiste en disminuir o eliminar la tumoración y las complicaciones por fotocoagulación con laser, crioterapia, radiación, terapia fotodinámica, uso de antiangiogénicos y cirugía [3-10], la eficacia del tratamiento depende de la localización del angioma, tiene éxito en los localizados en la periferia de la retina, en los localizados encima de la papila ocasiona disminución de la visión [5,6]. Los angiomas de retina son esporádicas; también pueden ser hereditarias en la enfermedad de von Hippel-Lindau (eVHL).
La eVHL se caracteriza por desarrollar varios tumores, benignos y malignos que afectan el sistema nervioso central (hemangioblastoma), la retina (angioma), el riñon (carcinoma de células claras), glándula suprarrenal (feocromocitoma), pancreas, epidídimo (quistes), entre otros [11,12]. La eVHL se hereda de manera autosómica dominante, con penetrancia del 90%, es causada por una mutación en el gen VHL ubicado en el brazo corto del cromosoma 3 (3p25) que da por resultado la pérdida de la proteína supresora de tumor VHL, cuya función principal es la regulación negativa del factor de crecimiento vascular endotelial (VSGF), lo que conduce a la presencia de múltiples tumores [13,14]. En USA y el este de Inglaterra la incidencia de esta enfermedad es de 1 en 36,000 nacidos vivos; la prevalencia es de 1 en 39,000 en el suroeste de Alemania y en el este de Inglaterra es de 1 en 53,000 [15,16]. El hemangioblastoma de sistema nervioso central (SNC) se presenta en el 60-80% de pacientes con eVHL, la localización más frecuente es en cerebelo y medula espinal [17-19]. El 50% de pacientes con angioma de retina, el 50% con feocromocitoma y el 1% con carcinoma renal de células claras cursan con eVHL [20]. La clasificación clínica de eVHL considera dos grupos: eVHL tipo 1, sin feocromocitoma y eVHL tipo 2, con feocromocitoma, este tipo se subdivide en 2A cuando hay cáncer en riñon, 2B sin cáncer de riñon y 2C solo presentan feocromocitoma [21,22]. En ausencia de historia familiar de eVHL el diagnóstico se basa en la presencia de al menos dos hemangioblastomas en SNC, o bien un hemangioblastoma en SNC y otra tumoración visceral asociada a eVHL. Cuando la historia familiar es positiva el diagnóstico se basa en la presencia de un tumor asociado a la eVHL (hemangioblastoma, feocromocitoma, angioma de retina, carcinoma renal de células claras, entre otros) [23]. El hemangioblastoma de SNC, frecuentemente es la primera manifestación de eVHL, cuando es hereditario es más agresivo que el esporádico, pues tiene crecimiento más rápido, es multifocal y recidivante, de aquí la importancia de diferenciar el esporádico del asociado a eVHL [16,18,22] La eVHL es una enfermedad compleja que requiere un tratamiento interdisciplinario; los familiares de un paciente con eVHL deben ser sujetos a un protocolo de estudio clínico y de imagen para identificar oportunamente las lesiones [24].
El objetivo de este estudio fue conocer las características clínicas de los pacientes con angiomas de retina y su relación con eVHL vistos en un hospital de tercer nivel.
Material y Método
Se realizó un estudio restrospectivo, observacional, analítico y descriptivo de los pacientes vistos en la consulta de oftalmología de un hospital de concentración de tercer nivel de la ciudad de México de 2010 al 2015, con diagnóstico de angioma de retina. En cada caso, se recabó historia clínica, con énfasis en historia familiar de eVHL, se revisó imágen clínica y fluorangiográfica del fondo de ojo e imágen de resonancia magnética y ultrasonido de sistema nervioso central y abdomen.
Los pacientes dieron su consentimiento para participar en este estudio que fue aceptado por el Comité de Ética del hospital. Lactante reingresa dos meses después a centro hospitalario con diagnóstico de neumonía adquirida en la comunidad y egresada con evolución satisfactoria.
Resultados
Durante el período de estudio de 54,996 consultas de pacientes que acudieron por primera vez al servicio de oftalmología, se diagnosticaron seis casos de angioma de retina; cuatro fueron femeninos (67%) y dos masculinos (33%), con edades entre 9 y 50 años, promedio de edad de 36 años.
De los seis casos, en tres se presentó angioma retiniano en un solo ojo, el ojo derecho (OD) en dos pacientes y el ojo izquierdo (OI) en uno; en los otros tres el angioma retiniano se presentó en los dos ojos. En los nueve ojos el angioma fue tumor único, menor a un diametro papilar, en siete ojos la localización fue periférica, cuatro localizados en cuadrante temporal inferior, y tres en las regiones temporal superior, nasal superior y nasal inferior, en un ojo la localización fue epipapilar, un ojo en ptisis bulbi. La agudeza visual promedio fue de 20/30. (Tabla 1) De los seis casos; dos casos correspondieron a angioma de retina esporádico, fueron unioculares y no tratados (caso 2 y 5) y cuatro casos a eVHL (casos 1,3,4 y 6), clasificados como eVHL tipo 1. En tres casos de eVHL el angioma de retina se localizó en ambos ojos (casos 3,4 y 6), y en el otro caso la localización fue en solo un ojo; tres ojos fueron tratados por presentar exudación y sangrado, dos ojos con laser y antiangiogénico (casos 3 y 4) (Figura 1), y un ojo con crioablación y antiangiogénico (Figura 3). En los cuatro casos con eVHL, se encontró hemangioblastoma en sistéma nervioso central, tumor renal y quiste pancreático (Figura 2) en dos casos y en ningún caso se identificó la presencia de Feocromocitoma (Tabla 2). Un paciente falleció a consecuencia del hemangioblastoma en SNC.
| Paciente |
Sexo |
Edad |
Agudeza Visual |
Lesiones |
Afección ocular |
|
| OD |
OI |
OD |
OI |
|
| 1 |
F |
9 |
20/40 |
20/40 |
TS |
Sin lesiones |
Unilateral |
eVHL |
| 2 |
F |
50 |
20/40 |
20/30 |
TI |
Sin lesiones |
Unilateral |
Aislado |
| 3 |
F |
36 |
20/150 |
20/400 |
TI |
TI |
Bilateral |
eVHL |
| 4 |
M |
37 |
20/20 |
- |
TI |
Ptisisbulbi |
Bilateral |
eVHL |
| 5 |
F |
40 |
20/30 |
20/30 |
Sin lesiones |
NS |
Unilateral |
Aislado |
| 6 |
M |
43 |
20/30 |
20/30 |
Yuxtapapilar |
NI |
Bilateral |
eVHL |
OD: Ojo Derecho; OI: Ojo Izquierdo; TS: Temporal Superior; TI: Temporal Inferior; NS: Nasal Superior; NI: Nasal Inferior; eVHL: Enfermedad de Von Hippel-Lindau
Tabla 1: Datos clínicos de pacientes con angioma de retina.
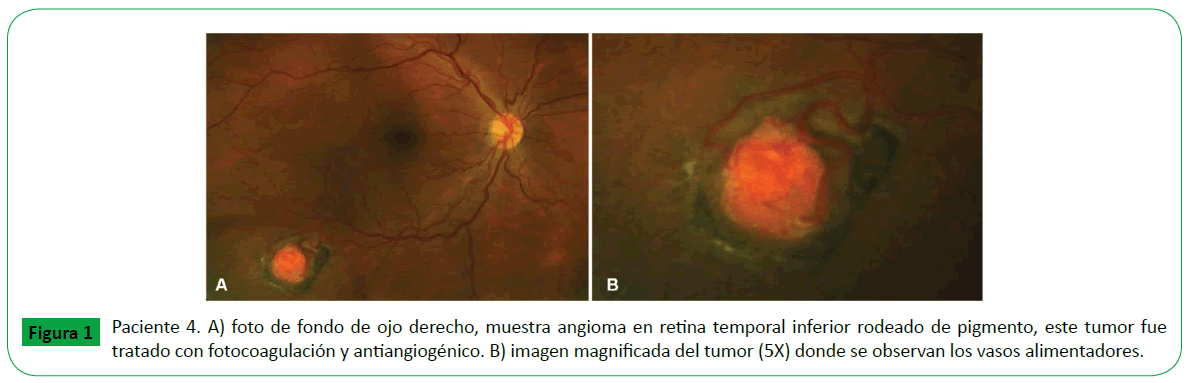
Figure 1: Paciente 4. A) foto de fondo de ojo derecho, muestra angioma en retina temporal inferior rodeado de pigmento, este tumor fue tratado con fotocoagulación y antiangiogénico. B) imagen magnificada del tumor (5X) donde se observan los vasos alimentadores.
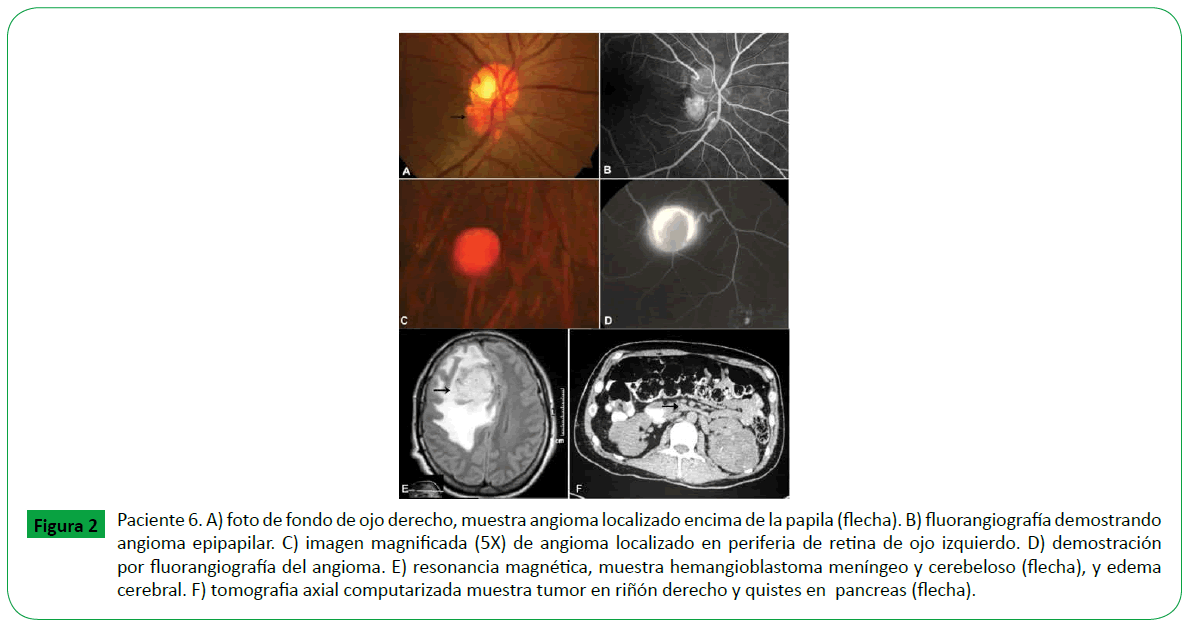
Figure 2: Paciente 6. A) foto de fondo de ojo derecho, muestra angioma localizado encima de la papila (flecha). B) fluorangiografía demostrando angioma epipapilar. C) imagen magnificada (5X) de angioma localizado en periferia de retina de ojo izquierdo. D) demostración por fluorangiografía del angioma. E) resonancia magnética, muestra hemangioblastoma meníngeo y cerebeloso (flecha), y edema cerebral. F) tomografia axial computarizada muestra tumor en riñón derecho y quistes en pancreas (flecha).
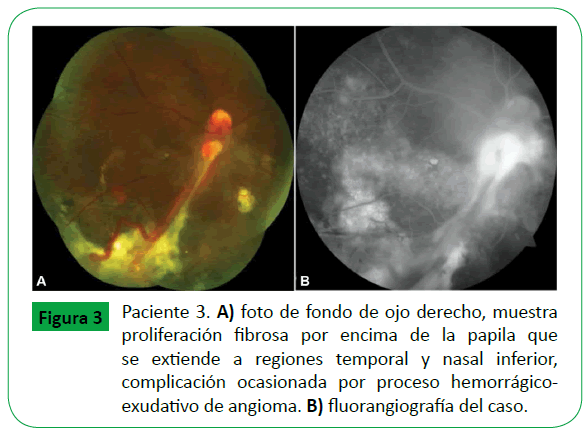
Figure 3: Paciente 3. A) foto de fondo de ojo derecho, muestra proliferación fibrosa por encima de la papila que se extiende a regiones temporal y nasal inferior, complicación ocasionada por proceso emorrágicoexudativo de angioma. B) fluorangiografía del caso.
| Paciente |
Sexo |
Edad |
Tumor SNC |
Quistepancreáticos |
Tumor renal |
Feocromocitoma |
| 1 |
F |
9 |
Si |
- |
- |
No |
| 3 |
F |
36 |
Si |
Si |
Si |
No |
| 4 |
M |
37 |
Si |
- |
- |
No |
| 6 |
M |
43 |
Si |
Si |
Si |
No |
Tabla 2: Datos clínicos de pacientes con enfermedad de Von Hippel-Lindau.
Discusión
En este estudio de angioma de retina el porcentaje de asociación con eVHL es similar al encontrado por otros autores [2,9,10]. El riesgo de pérdida de la visión se relacionó con el tamaño y localización del angioma; como la localización fue periférica en la mayoría de los casos y el tamaño menor a un diámetro papilar la capacidad visual fue de 20/30 ó mejor, Webster et al. [25] señalan que la pérdida de la visión se relaciona con el incremento de la edad, sin embargo, en cinco años de seguimiento de estos enfermos la visión no se modificó; anatómica y funcionalmente permanecieron estables, esto también fue observado por Toy et al. [26] En estos casos la agudeza visual fue mejor de 20/200 a diferencia del estudio de Chew [27] que indica que el 8% de los ojos con eVHL tienen una visión de 20/200 o inferior. En esta serie de casos como en otras los angiomas de retina periférica complicados con exudación y hemorragia son bien controlados con fotocoagulación, crioterapia y la aplicación de antiangiogénico en cavidad vitrea [24,28-31]. Walther et al. [32] señalan la presencia de feocromocitoma en 39% de los casos de eVHL, Welander y Pappaspyrou [33-35] encontraron en 30% de eVHL feocromocitomas extra-adrenales también llamados paragangliomas, en esta serie no se presentó ningún caso con estas patologías, Havekes [36] encontró que más del 70% de feocromocitomas en los niños son ocasionados por eVHL, en esta serie solo hubo un caso de un niño que no presentó feocromocitoma. La causa frecuente de muerte en la eVHL es el carcinoma renal, en estos casos solo un paciente murió por causa del hemangioblastoma de SNC. El hemangioblastoma de SNC fue la tumoración más frecuente después del angioma de retina, en los casos reportados, esto también ha sido señalado por otros autores [12,13,17].
Conclusión
Es importante la detección temprana de eVHL. Todos los pacientes con angioma de retina o hemangioblastoma de SNC, que son las manifestaciones más frecuentes de esta enfermedad, deben recibir una atención cuidadosa para detectar otras tumoraciones asociadas, y ofrecer una intervención multidisciplinaria oportuna.
10752
References
- Singh AD, Shields CL, Shields JA (2001) Von Hippel-Lindau disease. SurvOphthalmol46:117-142.
- Kreusel KM, Bechrakis NE, Krause L, Neumann HP, Foerster MH (2006) Retinal angiomatosis in von Hippel-Lindau disease: a longitudinal ophthalmologic study. Ophthalmology113:1418-1424.
- Wong WT, Chew EY (2008) Ocular Von Hippel-Lindau Disease: Clinical update and emerging treatments. CurrOpinOphthalmol19:213-219.
- Aiello LP, George DJ, Cahill MT, Wong JS, Cavallerano J, etal. (2002) Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology109:1745-1751.
- Madhusudan S, Deplanque G, Branbrooke JP, Casttell E, Taylor M, et al. (2004)Antiangiogenic therapy for von Hippel-Lindau disease. JAMA291:943-945.
- BuelowVM, Pape S, Hoerauf H (2007) Systemic bevacizumab treatment of a juxtapapillary retinal haemangioma. ActaOphthalmolScand85:114-116.
- Ziemssen F, Voelker M, Inhoffen W, Bartz-Schmidt KU, Gelisken F (2007) Combined treatment of a juxtapapillary retinal capillary hamangioma with intravitrealbevacizumab and photodynamic therapy. Eye21:1125-1126.
- Dahr SS, Cusick M, Rodriguez-Coleman H, Srivastava SK, Thompson DJ,et al. (2007)Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina27:150-158.
- Garcia-Arumi J, Sararols LH, Cavero L, Escalada F, Corcóstegui BF (2000) Therapeutic options for capillary papillary hemangiomas. Ophthalmology107:48-54.
- Kim H, Yi JH, Kwon HJ, Lee CS, Lee SC (2014) Therapeutic outcomes of retinal hemangioblastomas. Retina34:2479-2486.
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, et al. (2003) Von Hippel-Lindau disease. Lancet361:2059-2067.
- O´Brien FJ, Danapal M, Jairam S, Lalani AK, Cunningham J, et al. (2014) Manifestations of Von HippelLindau syndrome: a retrospective national review. QJMed107:291-296.
- Maher ER, Neumann HP, Richard S (2011) Von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet19:617-623.
- Gossage L, Eisen T, Maher ER (2015) VHL, the story of a tumor suppresor gene. Nat Rev Cancer15:55-64.
- Wittström E, Nordling M, Andréasson S (2014) Genotype-phenotype correlations, and retinal function and structure in von Hippel-Lindau disease. Ophthalmic Genet35:91-106.
- Decker J, Neuhaus C, Macdonald F, Branch H, Maher ER (2014) Clinical utility gene card for: von Hippel-Lindau. Eur J Hum Genet22:497-504.
- Bamps S, CalenberghVF, Vleeschouwer S, Loon VJ, Sciot R, et al. (2013)What the neurosurgeon should know about hemangioblastoma, both sporadic and in Von Hippel-Lindau disease: A literature review. SurgNeurolInt4:145-151.
- Bründl E, Schödel P, Ullrich OW, Brawanski A, Schebesch KM (2014)Surgical resection of sporadic and hereditary hemangioblastoma: Our 10-year experience and a literature review. SurgNeurolInt5:138-147.
- Wanebo JE, Lonser RR, Glen GM, Oldfield EH (2003)The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg98:82-94.
- Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, et al. (2002) Germ-line mutations in nonsyndromicPhaeochromocytoma. N Engl J Med346:1459-1466.
- DeLellis RA (2003) Pathology and molecular genetics of endocrine tumours. WHO cassification of tumours of endocrine organs. IARC press: Lyon.
- Gläsker S, Neumann HPH, Koch CA, Vortmeyer AO. Von Hippel- Lindau Syndrome. De Groot LJ, Beck-Peccoz P, Chrousos G, et al., editors. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000-2012
- Heo SJ, Lee CK, Hahnky, Kim G, Kim H, Choi SH, et al. A case of von Hippel-Lindau disease with colorectal adenocarcinoma, renal cell carcinoma and hemangioblastoma. Cancer Research and Treatment: Official Journal of Korean Cancer Association 2015
- Shanbhoque KP, Hoch M, Fatterpaker G, Chandarana H (2016) Von Hippel-Lindau Disease: Review of Genetics and Imaging. RadiolClin North Am54:409-422.
- Webster Ar, Maher ER, Moore AT (1999) Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutations. Arch Ophthalmol1173:371-378.
- Toy BC, Agrón E, Nigam D, Chew EY, Wong WT (2012)Longitudinal analysis of retinal hemangioblastomatosis and visual function in ocular von Hippel-Lindau disease. Ophthalmology 119:2622-2630.
- Chew EY (2005) Ocular manifestations of von Hippel-Lindau disease. Clinical and genetic investigations. Trans Am OphthalmolSoc103:495-511.
- Gaudric A, Krivosic V, Duguid G, Massin P, Girand S, Richard S (2011)Vitreoretinal surgery for severe retinal capillary hemangiomas in von Hippel-Lindau disease. Ophthalmology118:142-149.
- Singh AD, NouriM, Shields CL, Shields JA, Perez N (2002) Treatment of retinal capillary hemangioma. Ophthalmology109:1799-1806.
- Schmidt D, Natt E, Neumann HP (2000) Long-term results of laser tratment for retinal angiomatosis in von Hippel-Lindau disease. Eur J Med Res 5:47-58.
- Ach T, Thiemeyer D, Hoeh AE, Schaal KB, Dithmar S (2010)Intravitrealbevacizumab for retinal capillary hemangioma: longterm results. ActaOphthalmol88:137-138.
- Walther MM, Reiter R, Keiser HR (1999) Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochrocytoma gives insight into natural history of pheochromocyroma. J Urol162:659-664.
- Hulsteijn LT, Louisse A, Havekes B, Kaptein AA, Jansen JC, et al. (2013) Quality of life is decreased in patients with paragangliomas. Eur J Endocrinol168:689-697.
- Welander J, Soderkvist P, Gimm O (2011) Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. EndocrRelat Cancer18:253-276.
- Papaspyrou K, Mewes T, Rossmann H, Fottner C, Schneider-Raetzke B, et al. (2012) Head and neck paragangliomas: Report of 175 patients (1989-2010).Head Neck34:632-637.
- Havekes B, Romijn JA, Eisenhofer G, Adams K, Pacak K (2009) Update on pediatric pheochromocytoma. PediatrNephrol24:943-950.

