Introduction
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality throughout the world [1]. It is associated with progressive disability and functional impairment, and represents a major economic and social burden worldwide [2]. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines COPD as a disease characterized by progressive airflow limitation that is not fully reversible, associated with an abnormal inflammatory response of the lungs to noxious particles or gases [3]. Although COPD refers to a broad group of lung diseases characterized by airflow limitation, parenchymal destruction (emphysema) and small-airway obstruction (chronic bronchitis) are the most important phenotypes [4]. Emphysema, which is characterized by permanent inflammation and irreversible destruction of alveolar walls, leading to airspace enlargement, loss of elastic recoil and hyperinflation [5], is the most studied of these conditions.
Pathophysiology of COPD
COPD may be considered a complex disease, as several different mechanisms seem to be involved in its pathophysiology. The most accepted hypothesis is an imbalance between elastase and antielastase activity that leads to enzymatic degradation of elastin [6,7]. Such an imbalance is observed after long-term cigarette smoke (CS) exposure [8].
CS or other inhaled irritants activate epithelial cells to release growth factors, such as transforming growth factor beta (TGF-β) and fibroblast growth factor (FGF), which induce fibroblast proliferation, resulting in small-airway inflammation and fibrosis [9,10]. Simultaneously, macrophages are activated and release proinflammatory cytokines, such as tumor necrosis factor- alpha (TNF-α), interleukin (IL)-1β, and IL-6, which amplify lung inflammation and contribute to disease progression. Macrophages also release several chemokines that attract circulating cells into the lungs, such as CCL 2 (an attractant of monocytes, which later differentiate into macrophages within lung tissue), CXCL8 (an attractant of neutrophils, which can also be recruited by LTB-4), and CXCL-9, CXCL-10, and CXCL-11 (attractants of Th1 cells, which release interferon gamma [IFN-γ]) [8,10]. Many of the inflammatory genes overexpressed in COPD smokers are governed by upregulation of the transcription factor nuclear NF-κB [11]. In non-stimulated cells, NF-κB is found in alveolar macrophages and airway cells in an inactive non-DNA binding form, associated with its inhibitory protein κBα (IκBα) [12]. However, cigarette smoke promotes IκBα degradation, which unmasks NF-κB, allowing it to migrate to the nucleus, bind to DNA and initiate gene transcription [13]. NF-κB is a heterodimer composed of two subunits, p65 and p50; the former is increased in bronchial biopsy specimens [14] and sputum [15] from smokers as compared to nonsmokers.
Macrophages and neutrophils contribute to persistent inflammation and to an oxidant/antioxidant imbalance by releasing reactive oxygen species (ROS) that induce epithelial and endothelial cell apoptosis and inactivate antiproteolytic defense mechanisms, such as tissue inhibitors of metalloproteases (TIMPs) and alpha-1 antitrypsin (α1-AT) [16,17]. Moreover, the oxidative stress can reduce levels of nuclear factor (erythroid-derived 2)- like 2 (Nrf2) [18-20], which regulates the cellular antioxidant response by upregulating genes involved in augmenting cellular antioxidant capacity and promotes expression of genes involved in detoxification of ROS and electrophilic compounds [21,22]. Thus, low levels of Nrf2 are associated with increased susceptibility to neutrophilic inflammation [23] and histone deacetylase-2 (HDAC2) degradation [24]. HDAC2 is an epigenetic regulator and a critical component of the corticosteroid receptor complex that mediates repression of NF-κB transcriptional activity by deacetylating histones in proinflammatory gene promoters [25] and deacetylating the corticosteroid receptor [26]. Its levels are substantially reduced in the alveolar macrophages and distal structural cells of patients with COPD, which is associated with increased disease severity and airway inflammation [27] and promotion of corticosteroid resistance [25,28]. Thus, the disruption of epigenetic mechanisms has important implications to disease progression. Apoptosis is also related to the release of granzymes and perforins by CD8+ lymphocytes [29] and to low levels of vascular endothelial growth factor (VEGF) as a result of the endothelial cell destruction process [30].
Matrix metalloproteinases (such as MMP-8, MMP-9, and MMP- 12) released by macrophages and neutrophils can degrade a variety of matrix components, including collagen and elastin, leading to alveolar wall destruction [31,32]. Subsequently, elastin fragments become chemoattractants of further inflammatory cell influx [17]. The abnormal collagen remodeling that follows plays a major role both in COPD progression [33,34] and in lung function [35]. Figure 1 illustrates the above concepts related to COPD pathophysiology.
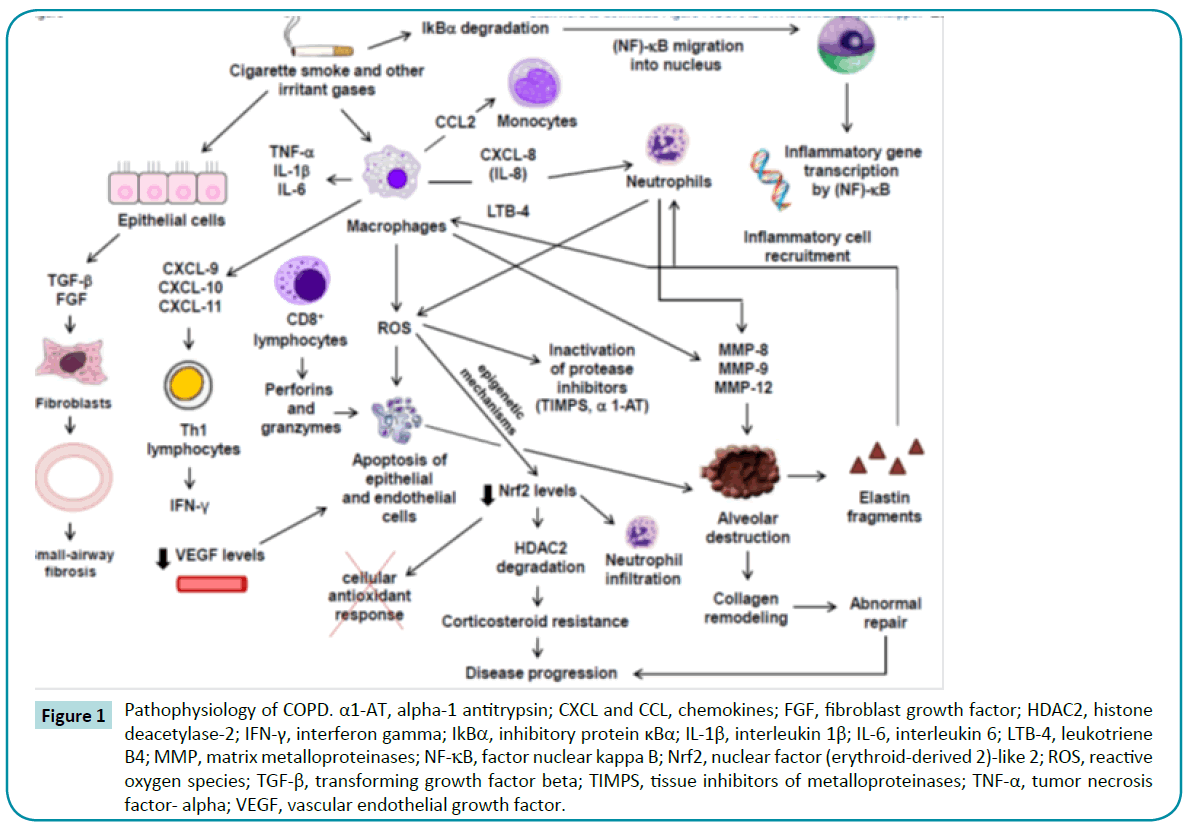
Figure 1: Pathophysiology of COPD. α1-AT, alpha-1 antitrypsin; CXCL and CCL, chemokines; FGF, fibroblast growth factor; HDAC2, histone deacetylase-2; IFN-γ, interferon gamma; IkBα, inhibitory protein κBα; IL-1β, interleukin 1β; IL-6, interleukin 6; LTB-4, leukotriene B4; MMP, matrix metalloproteinases; NF-κB, factor nuclear kappa B; Nrf2, nuclear factor (erythroid-derived 2)-like 2; ROS, reactive oxygen species; TGF-β, transforming growth factor beta; TIMPS, tissue inhibitors of metalloproteinases; TNF-α, tumor necrosis factor- alpha; VEGF, vascular endothelial growth factor.
Although COPD affects primarily the lungs, it is known to be associated with systemic effects [36]. The major comorbidities observed in COPD are: cardiovascular impairment, such as pulmonary hypertension and heart failure [37,38] diaphragmatic muscle dysfunction [39] skeletal muscle wasting [40] osteoporosis; diabetes [41] and malnutrition and weight loss [42,43]. These comorbidities reduce patient quality of life [43] and increase the risk of exacerbations [44] and mortality [45]. In addition, respiratory infections can worsen these comorbidities and compound their impact on the patient’s life [43].
Major Experimental Models of COPD
There are three major experimental approaches for induction of emphysema: CS exposure, elastase instillation, and genetic manipulation.
Cigarette Smoke Exposure
The severity of emphysema induced by the CS exposure model can be influenced by some factors, such as differences in animal strains, smoke concentration, and sex. Vecchio et al. [46] showed that C57BL/6 mice exhibited higher levels of proinflammatory cytokines, ROS, and MMPs, with lower levels of glutathione peroxidase (GPX), than Institute of Cancer Research (ICR) mice. Smoke concentration also plays an important role. Hodge-Bell et al. exposed male C57 and ICR mice to nose-only CS inhalation from 2R4F reference cigarettes, at concentrations of 75, 250, and 600 micrograms of total particulate matter (TPM) per liter. They observed an approximately 13% increase in mean linear intercept (Lm) only in mice exposed to 600 mcg TPM/L [47]. Regarding sex, March et al. showed that female A/J mice develop emphysema earlier than male mice from the same strain [48]. CS studies have also been done in rats [49-54]. CS exposure produces not only pulmonary alterations, but also systemic manifestations, such as weight loss [55-59], oxidative modifications of muscle protein in respiratory and limb muscles [60], reduction of skeletal muscle strength and increase in catabolic factors [59], systemic inflammation [61], and pulmonary arterial hypertension [62] (Table 1). On the other hand, most models cannot reproduce the features of severe emphysema as observed in humans, which would translate into GOLD stages 3 or 4. Usually, only mild features (corresponding to GOLD stages 1 or 2) are observed, regardless of exposure time [63], whereas in humans, the majority of morbidity and mortality occurs in patients with severe disease [64]. Moreover, all changes induced by CS exposure take time to be observed [61,65,66]. Finally, unlike in human advanced COPD, the lesions induced by this model do not progress after cessation of CS exposure. Jobse et al. recently showed that, although CS exposure resulted in an increase in mononuclear cells and neutrophils, airspace enlargement, and V/Q mismatch, inflammatory cell levels returned to control values and V/Q parameters returned to normal after cessation of exposure [67].
| Study |
Strain and Species |
Interventions |
Outcomes |
| Gosker et al., 2009 [55] |
C57BL/6 mice |
Animals exposed to CS 5 days/week for 6 months |
Pulmonary inflammation, weight loss, and reduction in type IIA oxidative fiber proportion in the soleus muscle |
| Tang et al., 2010 [56] |
C57BL/6 mice |
Animals exposed to CS daily for 8 or 16 weeks |
Weight loss, reduction in citrate synthase and beta-hydroxyacyl CoA dehydrogenase in the soleus muscle |
| Tomoda et al., 2012 [57] |
Wistar rats |
Animals exposed to CS twice a day, 5 days/week for 4 weeks |
Food intake, weight gain, and plasma levels of leptin reduced |
| Esquivel et al., 2014 [58] |
Wistar rats |
Animals exposed to CS 20 times/day, 5 days/week for 2 months |
Body weight, abdominal fat, and plasma levels of leptin reduced |
| Kamiide et al., 2015 [59] |
Wistar rats |
Animals exposed to CS for 12 weeks |
Body weight, food intake, and skeletal muscle strength reduced; mRNA expression of catabolic factors increased |
| Barreiro et al., 2012 [60] |
AKR/J mice |
Animals exposed to CS for 6 months |
Weight gain reduced, oxidative stress levels in diaphragm and gastrocnemius increased. Proteins involved in glycolysis, ATP production and distribution, carbon dioxide hydration, and muscle contraction were carbonylated in respiratory and limb muscles |
| Krüger et al., 2015 [61] |
C57BL/6J mice |
Animals exposed to CS for 8, 16, 24, and 32 weeks |
Reduced body mass, time-dependent decrease in muscle mass, type I oxidative fibers, and muscle cross-sectional area |
| Braber et al., 2010 [62] |
A/J mice |
Animals exposed to CS 5 days/week for 20 weeks |
Right ventricle hypertrophy |
Table 1: Models of emphysema induced by cigarette smoke (CS).
Elastase Instillation
Porcine pancreatic elastase (PPE) offers the advantages of being inexpensive and able to induce features of panacinar emphysema [64,68,69] and more widespread lung damage [17]. In one study of C57BL/6 mice, intratracheal elastase administration produced a greater amount of low-density areas than CS exposure, as observed on quantitative micro-computed tomography [70]. A wide variety of studies have highlighted the proteolytic activity of elastase in causing structural changes, such as higher mean linear intercept and alveolar enlargement both in mice [71-75] and in rats [76-79] (Table 2). Furthermore, several studies reported changes in ECM composition after elastase administration, such as disorganized elastin [80,81], degradation of proteoglycans [82], and abnormal collagen remodeling [83-88]. However, as in CS models, these effects are dependent on several factors, including strain; enzyme dose at each instillation; and number of elastase challenges (Figure 2). Limjunyawong et al. recently reported that BALB/C mice are more sensitive than C57BL/6J to elastase injury, as demonstrated by significantly greater mortality, weight loss, decline in lung function, and loss of alveolar tissue [89]. Regarding dose and number of elastase challenges, Lüthje et al. demonstrated that mice subjected to five elastase administrations with a 1-week interval between them developed not only a more severe alveolar destruction, but also systemic manifestations, such as weight loss, diaphragmatic dysfunction, exercise intolerance, and pulmonary arterial hypertension. Unlike with the nonprogressive CS exposure model, these changes persisted for 6 months after injury induction [90]. Similar results have been demonstrated by other groups [74,81,82,91-94]. Furthermore, after multiple elastase instillations in mice, Cruz et al. and Antunes et al. reported the development of pulmonary arterial hypertension and cor pulmonale, one of the leading causes of death in human patients with emphysema [95,96].
| Study |
Strain and Species |
Interventions |
Outcomes |
| Takahashi et al., 2008 [72] |
C57BL/6 mice |
Single intratracheal injection of elastase |
Higher mean linear intercept and alveolar epithelial damage |
| Harada et al., 2009 [73] |
C57BL/6J mice |
Single intratracheal injection of elastase |
Alveolar enlargement, increased lung volume, upregulation of lung dendritic cells |
| Moreno et al., 2014 [74] |
C57BL/6 mice |
Single intratracheal injection of elastase |
Alveolar enlargement, increased levels of matrix metalloproteinases, degradation of fibronectin |
| Santos et al., 2014 [75] |
Swiss mice |
Single intratracheal injection of elastase |
Higher mean linear intercept, alveolar enlargement, reduced lung elastance |
| Onclinx et al., 2006 [76] |
Sprague-Dawley rats |
One or two intratracheal injections of elastase |
Alveolar enlargement, increased dynamic compliance |
| Furuya et al., 2012 [77] |
Wistar rats |
Single intratracheal injection of elastase |
Alveolar enlargement, reduced arterial oxygen tension |
Bianchi et al., 2015 [78]
Boiati et al., 2010 [79] |
Wistar rats |
Single intratracheal injection of elastase |
Higher mean linear intercept |
Table 2: Models of emphysema induced by elastase.
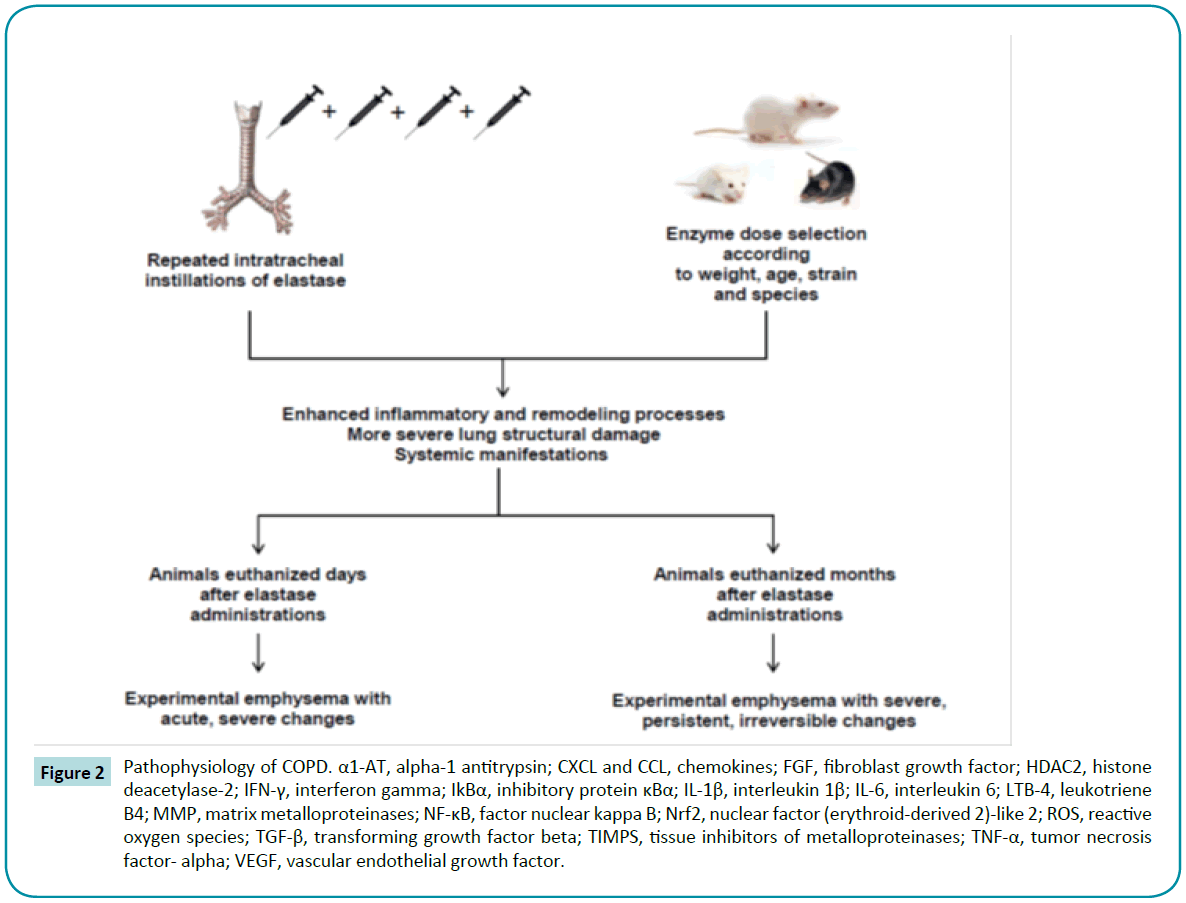
Figure 2: Pathophysiology of COPD. α1-AT, alpha-1 antitrypsin; CXCL and CCL, chemokines; FGF, fibroblast growth factor; HDAC2, histone deacetylase-2; IFN-γ, interferon gamma; IkBα, inhibitory protein κBα; IL-1β, interleukin 1β; IL-6, interleukin 6; LTB-4, leukotriene B4; MMP, matrix metalloproteinases; NF-κB, factor nuclear kappa B; Nrf2, nuclear factor (erythroid-derived 2)-like 2; ROS, reactive oxygen species; TGF-β, transforming growth factor beta; TIMPS, tissue inhibitors of metalloproteinases; TNF-α, tumor necrosis factor- alpha; VEGF, vascular endothelial growth factor.
Genetic Manipulation
With the advent of modern molecular biology techniques, transgenic mice can now be bred with artificially introduced alterations in their genome resulting in overexpression (“gain of function”) or low expression (“loss of function”) of the gene of interest. Gain-of-function models have added further proof to the protease-antiprotease imbalance hypothesis [97]. In this line, mice overexpressing human interstitial collagenase (MMP- 1) showed airspace enlargement [98] and degradation of type III collagen [99,100]. Furthermore, in mice, Wang et al. have demonstrated that overexpression of interferon gamma (IFN-γ) caused prominent protease and antiprotease alterations, such as induction and activation of MMP-12, MMP-9 and cathepsins B, H, D, S, and L, and the selective inhibition of secretory leukocyte proteinase inhibitor [101].
Targeted mutagenesis also allows investigators to generate strains of COPD mice that lack individual proteins (loss-offunction mutations), such as elastin [102], fibulin-5 [103], platelet derived growth factor (PDGF) [104,105], fibroblast growth factor receptor (FGFR) [106], surfactant protein-D (SP-D) [107-109], tissue inhibitor of metalloproteinases-3 (TIMP-3) [110], and ATP binding cassette A3 (Abca3), a lipid transport protein required for synthesis and storage of pulmonary surfactant in type II cells in the alveoli [111]. Recently, Holm et al. developed a mouse model lacking microfibrillar-associated protein 4 (MFAP4), an important protein localized into elastic fibers in blood vessels and the interalveolar septa of the lungs. These mice exhibit increased total lung capacity and evidence of a decrease in mean density of the lung parenchyma on breath-hold gated microcomputed tomography (micro-TC) [112]. Rangasamy et al. showed that disruption of the Nrf2 gene led to earlier onset and more extensive features of emphysema in mice exposed to CS, suggesting that this gene determines susceptibility to lung inflammation, oxidative stress, and alveolar cell apoptosis [23]. Similarly, Adenuga et al., using Nrf2 and HDAC2 knockout mice, observed that HDAC2 deficiency led to an increase in CS-induced lung inflammation and steroid resistance [24] (Table 3).
| Study |
Strain |
Interventions |
Outcomes |
| Wendel et al., 2000 [102] |
C57BL/6 mice |
Animals lacking elastin (Eln -/-) |
Dilated and few air sacs, impaired airway branching, attenuated tissue septae |
| Nakamura et al., 2002 [103] |
C57BL/6 mice |
Animals lacking fibulin-5 (fibulin-5-/-) |
Disorganized elastic fiber system throughout the body, characterized by tortuous aorta with loss of compliance, severe emphysema, and cutis laxa |
| Boström et al., 1996[104] |
C57BL/6 mice |
Animals lacking platelet-derived growth factor A (PDGF-A-/-) |
Alveolar hyperinflation, abnormally large and air-filled cavities, failure of alveolar septation, loss of alveolar myofibroblasts, corpulmonale |
| Lindahl et al., 1997[105] |
C57BL/6 mice |
Animals lacking platelet-derived growth factor A (PDGF-A-/-) |
Reduced deposition of elastin fibers in the lung parenchyma, development of early lung emphysema due to complete failure of alveogenesis |
| Weinstein et al., 1998 [106] |
C57BL/6 mice |
Animals lacking fibroblast growth factor receptors 3 and 4 (FGFR-3-/-, FGFR-4-/-) |
Failure of alveogenesis, reduced elastin synthesis, bronchopulmonary dysplasia, growth retardation |
Wert et al., 2000 [107]
Yoshida et al., 2001 [108]
Knudsen et al., 2009 [109] |
C57BL/6 mice |
Animals lacking surfactant protein-D (SP-D -/-) |
Postnatal airspace enlargement, subpleural fibrosis, chronic inflammation with infiltration of foamy alveolar macrophages, increased MMP activity and oxidant production by alveolar macrophages |
| Leco et al., 2001 [110] |
C57BL/6 mice |
Animals lacking tissue inhibitor of metalloproteinases-3 (TIMP-3 -/-) |
Spontaneous air space enlargement, impaired lung function, enhanced destruction of extracellular matrix molecules |
| Besnard et al., 2010 [111] |
C57BL/6 mice |
Animals lacking ATP-binding cassette A3 (Abca3 -/-) |
Death shortly after birth from respiratory distress related to surfactant deficiency. Surviving mice exhibited decreased expression of mRNAs associated with lipid synthesis and fewer lamellar bodies |
| Holm et al., 2015 [112] |
C57BL/6 mice |
Animals lacking microfibrillar-associated protein 4 (Mfap-4 -/-) |
Increased lung volumes and compliance, alveolar hyperinflation |
| Rangasamy et al., 2004 [23] |
C57BL/6 mice |
Animals lacking nuclear factor (erythroid-derived 2)-like 2 (Nrf-2 -/-) and exposed to CS for 6 months |
Increased apoptotic alveolar septal cell (endothelial and type II epithelial cell) counts, oxidative stress, bronchoalveolar inflammation |
| Adenuga et al., 2010 [24] |
C57BL/6J mice |
Animals lacking nuclear factor (erythroid-derived 2)-like 2 (Nrf-2 -/-) and deacetylase 2 (HDAC-2 -/-) exposed to CS for 3 days and later treated with budesonide |
Neutrophil influx in the lungs and BALF, increased levels of KC and MCP-1 in BALF, steroid insensitivity |
Table 3: Loss-of-function knockout mouse models.
In some mouse strains, spontaneous development of emphysema has been observed in association with genetic abnormalities. Pallid [7,113,114] and tight skin [115,116] mice have α1-antitrypsin deficiency, which results in reduced elastase inhibitory capacity, leading to emphysematous lesions at the age of 2–4 weeks.
In short, gene-targeting techniques are very useful tools to identify the role of distinct genes in the regulation of pulmonary homeostasis and to examine potential mechanisms underlying human COPD [117]. However, a major disadvantage of these models is that the gene of interest is also expressed in other organs [97], which can cause systemic effects.
COPD Exacerbations
Frequent acute exacerbations are observed during the life course of COPD patients [118]. Most exacerbations are triggered by infection, usually viral [119] or bacterial [120]. Repeated exacerbations are associated with worse prognosis [121,122], airway inflammation [123], and lung function impairment [124] (Figure 3).
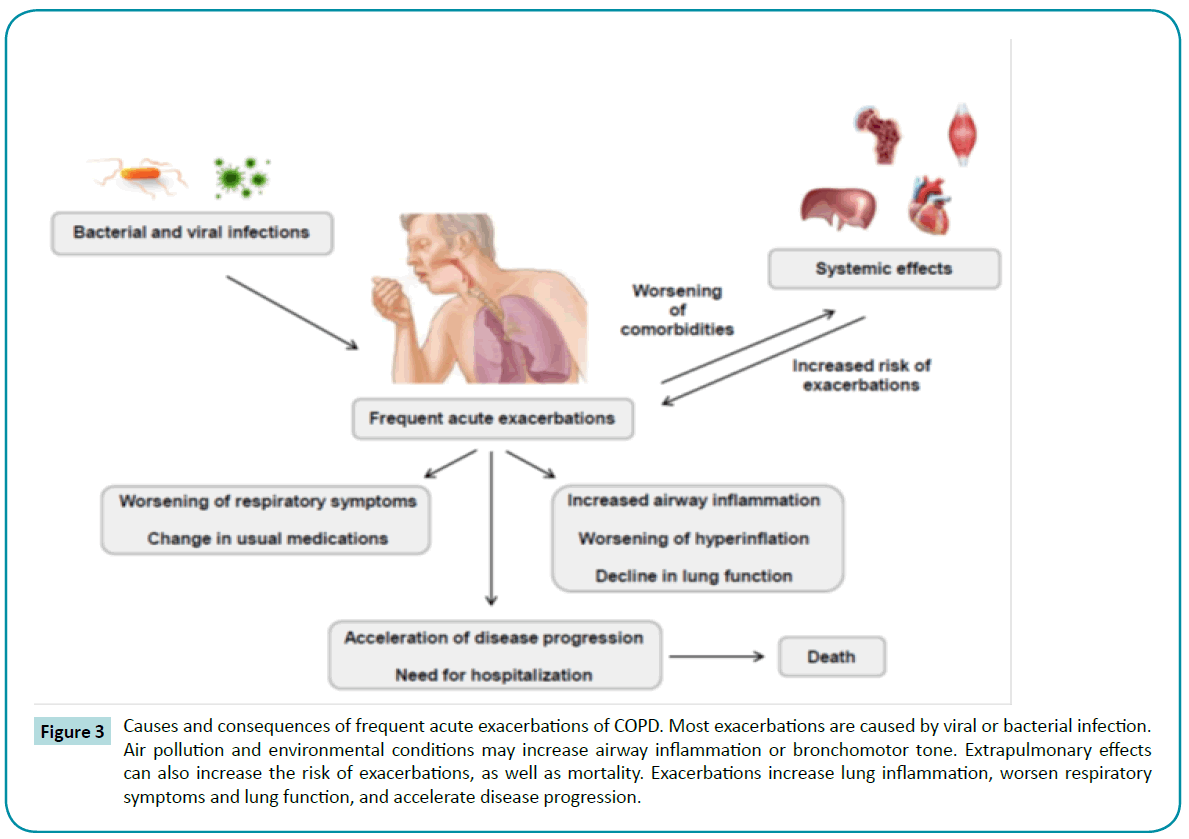
Figure 3: Causes and consequences of frequent acute exacerbations of COPD. Most exacerbations are caused by viral or bacterial infection. Air pollution and environmental conditions may increase airway inflammation or bronchomotor tone. Extrapulmonary effects can also increase the risk of exacerbations, as well as mortality. Exacerbations increase lung inflammation, worsen respiratory symptoms and lung function, and accelerate disease progression.
The susceptibility of smokers with COPD to respiratory infections is greater than that of nonsmokers, because CS exposure causes several disruptions to the innate lung defenses, such as impairment of mucociliary clearance [125], reductions in ciliary beat frequency and in the numbers of ciliated cells due to squamous metaplasia [126], reduction in the concentrations of surfactant proteins A and D [127], salivary lysozyme and sputum secretory leukocyte protease inhibitor deficiency [128,129], and impairment of phagocytosis by alveolar macrophages [130,131] (Figure 4). In addition, even a stable COPD condition is associated with respiratory pathogens in the airways, which worsens airflow conductance [132,133] and triggers an inflammatory response [15,123,134-136] and presence of inflammatory markers in sputum [133,137-139]. During exacerbations, these inflammatory cells and mediators increase [140,141], as does the H2O2 concentration in exhaled breath condensate [142,143].
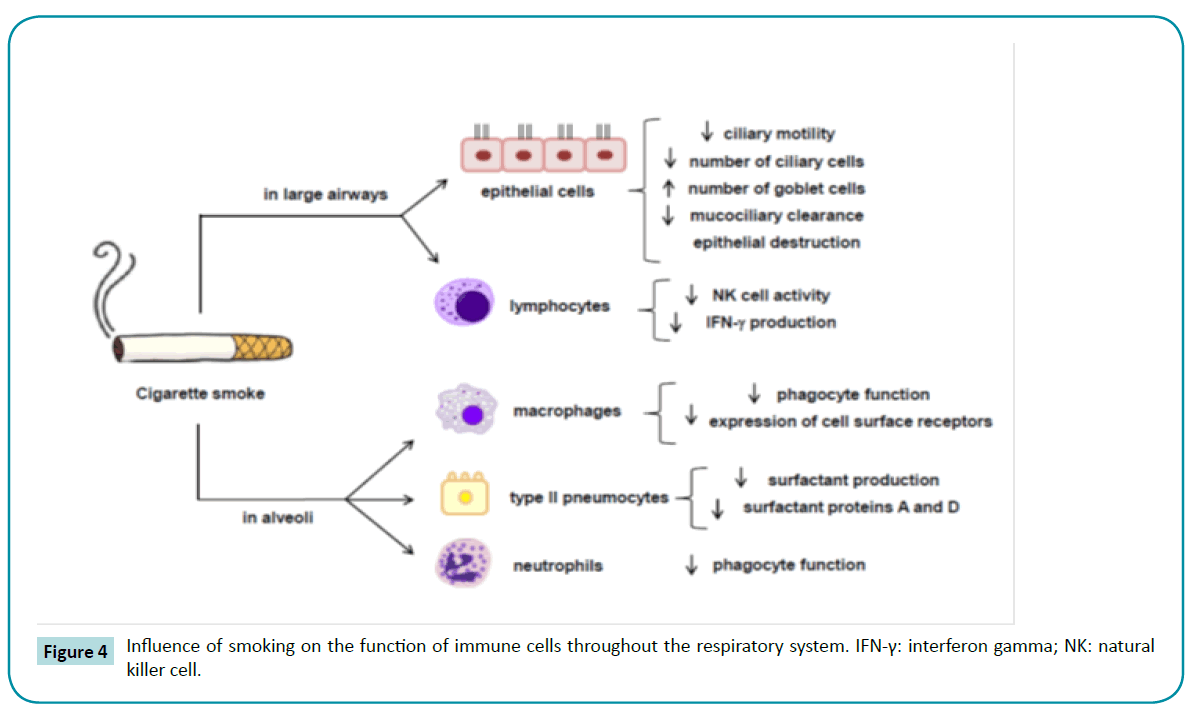
Figure 4: Influence of smoking on the function of immune cells throughout the respiratory system. IFN-γ: interferon gamma; NK: natural killer cell.
Although many acute COPD exacerbations can be treated on an out¬patient basis, some cause a greater decline in lung function, requiring hospitalization [124]. Therefore, effective strategies to reduce the incidence of COPD exacerbations and their duration are needed. The administration of corticosteroids has long been a mainstay of therapy for acute COPD exacerbations; however, chronic corticosteroid use is associated with many adverse events [144] and with increased risk of pneumonia, possibly due to their immunosuppressive action [145-147]. Additionally, inhaled or systemic corticosteroids fail to attenuate chronic inflammation in some patients, due to increased oxidative stress [148,149]. Corticosteroids suppress inflammation by recruiting HDAC2 to NF-κB-driven pro-inflammatory gene promoters, thereby inhibiting the transcription of these genes [150]. Thus, the inability of corticosteroids to suppress inflammation in some cases of COPD can be associated with loss of HDAC2 activity [151]. Several studies about the use of specific antibiotics in COPD exacerbations have been conducted [152-154], and it is known that antibiotic therapy can reduce sputum purulence [155]. On the other hand, such therapy may become ineffective against resistant bacterial strains, especially when treatment is provided in-hospital [156,157].
As previously reported, animal models with different purposes have been developed to elucidate the pathophysiology of COPD exacerbations. There are two major experimental approaches to induction of COPD exacerbations: 1) intratracheal instillation of bacterial lipopolysaccharides (LPS) and 2) challenge with specific bacterial or viral strains.
Experimental Models of COPD Exacerbation
LPS Infection
Using mice, Stolk et al. induced baseline emphysema through elastase administration and later induced exacerbations with intratracheal instillations of LPS (500 μg in 200 μL saline) twice a week for up to 5 weeks. Six months later, a severe bronchial mucus cell hyperplasia and persistent increase in mean linear intercept, indicating irreversible tissue destruction, were observed [158]. Recently, Kobayashi et al. induced exacerbations using only a single intratracheal administration of LPS (1 mg/kg) to mice with Using mice, Stolk et al. induced baseline emphysema through elastase administration and later induced exacerbations with intratracheal instillations of LPS (500 μg in 200 μL saline) twice a week for up to 5 weeks. Six months later, a severe bronchial mucus cell hyperplasia and persistent increase in mean linear intercept, indicating irreversible tissue destruction, were observed [158]. Recently, Kobayashi et al. induced exacerbations using only a single intratracheal administration of LPS (1 mg/kg) to mice with elastase-induced emphysema. Three days after LPS treatment, the authors observed neutrophil infiltration and CD8+ cells in bronchoalveolar lavage fluid (BALF), as well as increased levels of MMP-9 and TIMP-1, whereas after 12 weeks, they found severe alveolar destruction, using the parameter of low-attenuation area percentage (LAA%) on micro-computed X-ray tomography, suggesting intense acute inflammation and severe, irreversible alveolar destruction, respectively [159].
In rats, a single massive LPS insult (40 mg/kg, intratracheally) has been reported to cause an inflammatory response followed by mucus hypersecretion and bronchoconstriction, which reproduces symptoms of an exacerbation [160]. Hardaker et al. reported in a CS model that aerosolized LPS (0.3 mg/mL) was able to increase neutrophil infiltration, mucus, and edema in the lungs and in BALF and to impair lung function [161]. Accordingly, in a CS model, a single LPS instillation (200 μg/kg, intratracheally) promoted inflammatory cell infiltration and remarkable gobletcell hyperplasia in tracheal, bronchial, and bronchiolar epithelium [162]. Recently, also in a CS model, Li et al. reported airspace enlargement, decreased expressions of surfactant protein (SP)-A and SP-C, and apoptosis of alveolar epithelial cells after two intratracheal LPS instillations (1 μg/μL) [163].
Bacterial Infection
Given that bacteria are the main cause of infections in COPD patients, the majority of studies use bacteria to induce exacerbations in emphysema models. In this line, Gashler et al. exposed C57BL/6 and BALB/c mice to CS for 8 weeks and subsequently challenged the animals with Haemophilus influenza (NTHI). In both strains, the authors observed an increase in pulmonary inflammation and lung damage. Furthermore, NTHI challenge led to prominent upregulation of some inflammatory mediators, such as monocyte chemotactic protein 1 (MCP-1), MCP-3, and MCP-5 [164]. Huvenne et al. investigated the effects of Staphylococcus aureus enterotoxin B (SEB) in C57BL/6 mice exposed to CS for 4 weeks. CD8+ T lymphocytes and granulocytes increased in BALF, and goblet cell hyperplasia was observed in the airway wall [165]. In an investigation of the impact of CS on bacterial clearance and immune inflammatory processes in mice, Drannik et al. found higher levels of TNF-α, IL-1β, IL-6, MCP-1, and MIP-2 in lung homogenates after acute P. aeruginosa infection [166]. Recently, Voss et al. also showed enhanced inflammation in the upper airways and lung tissue of C57BL/6 mice exposed to CS after colonization with NTHI and Streptococcus pneumoniae [167]. Similar results can be obtained after elastase-induced emphysema. In this line, Pang et al. showed decreased intercellular adhesion molecule 1 (ICAM-1) in airway epithelium and low NTHI clearance, with pathologic findings consistent with pneumonia, supporting the hypothesis that ICAM-1 promotes clearance of NTHI [168]. Furthermore, Wang et al. induced exacerbation through NTHI challenge after elastase-induced emphysema, and observed lung consolidation, capillary congestion, atelectasis, hemorrhage, neutrophil infiltration, and higher levels of TNF-γ and IL-8 in BALF and plasma [169] typical clinical signs of severe pneumonia. Finally, Ganesan et al. developed a more complex animal model in which they combined a bacterial source and LPS. The authors administered LPS and elastase simultaneously for four consecutive weeks in C57BL/6 mice. One week after the last exposure to LPS, animals were challenged with NTHI. Elastase/ LPS exposed mice exhibited delayed bacterial clearance with an increase in neutrophilic inflammation and prolonged mucus secretion as assessed by mucin gene expression and periodic acid–Schiff (PAS) staining of lung histology. Moreover, ex vivo macrophages showed deficient phagocytosis, possibly could be caused by decreased expression of scavenger receptor A [170]. Studies in rats are lacking.
Viral Infection
To date, very few studies have tried to induce viral infection to mimic COPD exacerbations. Bauer et al. exposed C57BL/6 mice to CS for 4 days and subsequently inoculated the animals with the influenza A(H1N1) virus. Mononuclear and neutrophil cells, MCP-1, MCP-3, and CXC (KC, MIP-2) chemokines increased in BALF [171]. Recently, using a single elastase treatment followed by rhinovirus (RV) infection in C57BL/6 mice, Singanayagam et al. reported increased airway neutrophilic and lymphocytic inflammation, increased expression of TNF-α and CXC motif chemokine 10 (CXCL10)/IP-10 (IFN-γ-induced protein 10), mucus hypersecretion, and preliminary evidence for increased airway hyperresponsiveness [172]. No major studies have been conducted in rats using viral infection.
Conclusions
Although no one animal model of COPD is able to mimic human disease, the different types of models available have provided valuable information not only about the complex and heterogeneous pathophysiology of this disease, but also about the mechanisms underlying its progression and exacerbations. A greater understanding of these mechanisms may help develop and test novel therapies to effectively prevent disease deterioration and minimize mortality.
7965
References
- Eisner MD, Anthonisen N, Coultas D, Kuenzli N, Perez-Padilla R, et al. (2010) An official American Thoracic Society public policy statement: Novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J RespirCrit Care Med 182: 693-718.
- Kim J, Lee TJ, Kim S, Lee E (2015) The economic burden of chronic obstructive pulmonary disease from 2004 to 2013.
- GOLD Global Strategy for Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease. Vancouver WA: Global Initiative for Chronic Obstructive Lung Disease2015.
- McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, et al. (2011) Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 365: 1567-1575.
- Snider GL (1989) Chronic obstructive pulmonary disease: a definition and implications of structural determinants of airflow obstruction for epidemiology. Am Rev Respir Dis 140: S3-S8.
- Snider GL (1986) Experimental studies on emphysema and chronic bronchial injury. Eur J Respir Dis Suppl 146: 17-35.
- Takubo Y, Guerassimov A, Ghezzo H, Triantafillopoulos A, Bates JH, et al. (2002) Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J RespirCrit Care Med 166: 1596-1603.
- MacNee W (2005) Pathogenesis of chronic obstructive pulmonary disease. Proc Am ThoracSoc 2: 258-266.
- Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, et al. (2001) Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J RespirCrit Care Med 164: 469-473.
- Barnes PJ (2008) The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest 118: 3546-3556.
- Christman JW, Sadikot RT, Blackwell TS (2000) The role of nuclear factor-kappa B in pulmonary diseases. Chest 117: 1482-1487.
- Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389: 349-352.
- Szulakowski P, Crowther AJ, Jiménez LA, Donaldson K, Mayer R, et al. (2006) The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J RespirCrit Care Med 174: 41-50.
- Di Stefano A, Caramori G, Oates T, Capelli A, Lusuardi M, et al. (2002) Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. EurRespir J 20: 556-563.
- Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, et al. (2003) Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 58: 348-351.
- Stockley RA, Mannino D, Barnes PJ (2009) Burden and pathogenesis of chronic obstructive pulmonary disease. Proc Am ThoracSoc 6: 524-526.
- Antunes MA, Rocco PR (2011) Elastase-induced pulmonary emphysema: insights from experimental models. An Acad Bras Cienc 83: 1385-1396.
- Goven D, Boutten A, Leçon-Malas V, Marchal-Sommé J, Amara N, et al. (2008) Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax 63: 916-924.
- Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, et al. (2008) Expression of concern: Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J RespirCrit Care Med 178: 592-604.
- Garbin U, FrattaPasini A, Stranieri C, Cominacini M, Pasini A, et al. (2009) Cigarette smoking blocks the protective expression of Nrf2/ARE pathway in peripheral mononuclear cells of young heavy smokers favouring inflammation. PLoS One 4:e8225-e8236.
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, et al. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. BiochemBiophys Res Commun236: 313-322.
- Jaiswal AK (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free RadicBiol Med 36: 1199-1207.
- Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, et al. (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114: 1248-1259.
- Adenuga D, Caito S, Yao H, Sundar IK, Hwang JW, et al. (2010) Nrf2 deficiency influences susceptibility to steroid resistance via HDAC2 reduction. BiochemBiophys Res Commun 403: 452-456.
- Barnes PJ (2009) Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol 71: 451-464.
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, et al. (2006) Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappa B suppression. J Exp Med 203: 7-13.
- Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, et al. (2004) Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med 200: 689-695.
- Malhotra D, Thimmulappa RK, Mercado N, Ito K, Kombairaju P, et al. (2011) Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest 121: 4289-4302.
- Gadgil A, Duncan SR (2008) Role of T-lymphocytes and pro-inflammatory mediators in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 3: 531-541.
- Morissette MC, Parent J, Milot J (2011) The emphysematous lung is abnormally sensitive to TRAIL-mediated apoptosis. Respir Res 12: 105.
- Janoff A (1985) Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. Am Rev Respir Dis 132: 417-433.
- Churg A, Zhou S, Wright JL (2012) Series "matrix metalloproteinases in lung health and disease": Matrix metalloproteinases in COPD. EurRespir J 39: 197-209.
- Laurent GJ (1991) Regulation of lung collagen production during wound healing. Chest 99: 67S-69S.
- Wright JL, Churg A (1995) Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. Am J Physiol 268: L17-L20.
- Suki B, Lutchen KR, Ingenito EP (2003) On the progressive nature of emphysema: roles of proteases, inflammation, and mechanical forces.Am J RespirCrit Care Med 168: 516-521.
- Choudhury G, Rabinovich R, MacNee W (2014) Comorbidities and systemic effects of chronic obstructive pulmonary disease. Clin Chest Med 35: 101-130.
- Bhatt SP, Dransfield MT (2013) Chronic obstructive pulmonary disease and cardiovascular disease. Transl Res 162: 237-251.
- Smith MC, Wrobel JP (2014) Epidemiology and clinical impact of major comorbidities in patients with COPD. Int J Chron Obstruct Pulmon Dis 9: 871-888.
- Ottenheijm CA, Heunks LM, Dekhuijzen RP (2008) Diaphragm adaptations in patients with COPD. Respir Res 9: 12.
- Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, et al. (2014) An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J RespirCrit Care Med 189: e15-e62.
- Barnes PJ, Celli BR (2009) Systemic manifestations and comorbidities of COPD. EurRespir J 33: 1165-1185.
- Agustí AG, Noguera A, Sauleda J, Sala E, Pons J, et al. (2003) Systemic effects of chronic obstructive pulmonary disease. EurRespir J 21: 347-360.
- Cavaillès A, Brinchault-Rabin G, Dixmier A, Goupil F, Gut-Gobert C, et al. (2013) Comorbidities of COPD. EurRespir Rev 22: 454-475.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, et al. (2005) Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax60: 925-931.
- McGarvey LP, Magder S, Burkhart D, Kesten S, Liu D, et al. (2012) Cause-specific mortality adjudication in the UPLIFT® COPD trial: findings and recommendations. Respir Med 106: 515-521.
- Vecchio D, Arezzini B, Pecorelli A, Valacchi G, Martorana PA, et al. (2010) Reactivity of mouse alveolar macrophages to cigarette smoke is strain dependent. Am J Physiol Lung Cell MolPhysiol 298: L704-L713.
- Hodge-Bell KC, Lee KM, Renne RA, Gideon KM, Harbo SJ, et al. (2007) Pulmonary inflammation in mice exposed to mainstream cigarette smoke. InhalToxicol 19: 361-376.
- March TH, Wilder JA, Esparza DC, Cossey PY, Blair LF, et al. (2006) Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. ToxicolSci 92: 545-559.
- Ning Y, Shang Y, Huang H, Zhang J, Dong Y, et al. (2013) Attenuation of cigarette smoke-induced airway mucus production by hydrogen-rich saline in rats. PLoS One 8: e83429.
- Li MC, Yang G, Zhou XD, Tselluyko S, Perelman JM. (2014) The pathophysiological mechanisms underlying mucus hypersecretion induced by cold temperatures in cigarette smoke-exposed rats. Int J Mol Med 33: 83-90.
- Zhou Y, Zhang Y, Guo Y, Zhang Y, Xu M, Het al. (2014) ß2-Adrenoceptor involved in smoking-induced airway mucus hypersecretion through ß-arrestin-dependent signaling. PLoS One 9: e97788.
- Zhou Y, Xu M, Zhang Y, Guo Y, Zhang Y, et al. (2014) Effects of long-term application of metoprolol and propranolol in a rat model of smoking. ClinExpPharmacolPhysiol 41: 708-715.
- Kamiide Y, Inomata N, Furuya M, Yada T(2015) Ghrelin ameliorates catabolic conditions and respiratory dysfunction in a chronic obstructive pulmonary disease model of chronic cigarette smoke-exposed rats. Eur J Pharmacol755:88-94.
- Wang W, Li X, Xu J (2015) Exposure to cigarette smoke downregulates β2-adrenergic receptor expression and upregulates inflammation in alveolar macrophages. InhalToxicol 27: 488-494.
- Gosker HR, Langen RC, Bracke KR, Joos GF, Brusselle GG, et al. (2009) Extrapulmonary manifestations of chronic obstructive pulmonary disease in a mouse model of chronic cigarette smoke exposure. Am J Respir Cell MolBiol 40: 710-716.
- Tang K, Wagner PD, Breen EC (2010) TNF-alpha-mediated reduction in PGC-1alpha may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol 222: 320-327.
- Tomoda K, Kubo K, Nishii Y, Yamamoto Y, Yoshikawa M, et al. (2012) Changes of ghrelin and leptin levels in plasma by cigarette smoke in rats. J ToxicolSci 37: 131-138.
- Esquivel AL, Pérez-Ramos J, Cisneros J, Herrera I, Rivera-Rosales R, et al. (2014) The effect of obesity and tobacco smoke exposure on inflammatory mediators and matrix metalloproteinases in rat model. ToxicolMech Methods 24: 633-643.
- Kamiide Y, Furuya M, Inomata N, Yada T (2015) Chronic exposure to cigarette smoke causes extrapulmonary abnormalities in rats. Environ ToxicolPharmacol 39: 864-870.
- Barreiro E, del Puerto-Nevado L, Puig-Vilanova E, Pérez-Rial S, Sánchez F, et al. (2012) Cigarette smoke-induced oxidative stress in skeletal muscles of mice. RespirPhysiolNeurobiol 182: 9-17.
- Krüger K, Dischereit G, Seimetz M, Wilhelm J, Weissmann N, et al. (2015) Time course of cigarette smoke-induced changes of systemic inflammation and muscle structure. Am J Physiol Lung Cell MolPhysiol 309: L119-L128.
- Braber S, Henricks PA, Nijkamp FP, Kraneveld AD, Folkerts G (2010) Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir Res 11: 99.
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, et al. (2007) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J RespirCrit Care Med 176: 532-555.
- Wright JL, Cosio M, Churg A (2008) Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell MolPhysiol 295: L1-15.
- Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, et al. (2011) IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One 6: e20333.
- Caron MA, Morissette MC, Thériault ME, Nikota JK, Stämpfli MR, et al. (2013) Alterations in skeletal muscle cell homeostasis in a mouse model of cigarette smoke exposure. PLoS One 8: e66433.
- Jobse BN, McCurry CA, Morissette MC, Rhem RG, Stämpfli MR, et al. (2014) Impact of inflammation, emphysema, and smoking cessation on V/Q in mouse models of lung obstruction. Respir Res 15: 42.
- Snider GL, Lucey EC, Stone PJ (1986) Animal models of emphysema. Am Rev Respir Dis 133: 149-169.
- Snider GL (1992) Emphysema: the first two centuries--and beyond. A historical overview, with suggestions for future research: Part 2. Am Rev Respir Dis 146: 1615-1622.
- Sasaki M, Chubachi S, Kameyama N, Sato M, Haraguchi M, et al. (2015) Evaluation of cigarette smoke-induced emphysema in mice using quantitative micro-computed tomography. Am J Physiol Lung Cell MolPhysiol 308: L1039-L1045.
- Parameswaran H, Majumdar A, Ito S, Alencar AM, Suki B (2006) Quantitative characterization of airspace enlargement in emphysema. J ApplPhysiol 100: 186-193.
- Takahashi S, Nakamura H, Seki M, Shiraishi Y, Yamamoto M, et al. (2008) Reversal of elastase-induced pulmonary emphysema and promotion of alveolar epithelial cell proliferation by simvastatin in mice. Am J Physiol Lung Cell MolPhysiol 294: L882-890.
- Harada H, Imamura M, Okunishi K, Nakagome K, Matsumoto T, et al. (2009) Upregulation of lung dendritic cell functions in elastase-induced emphysema. Int Arch Allergy Immunol 149 Suppl 1: 25-30.
- Moreno JA, Ortega-Gomez A, Rubio-Navarro A, Louedec L, Ho-Tin-Noé B, et al. (2014) High-density lipoproteins potentiate α1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am J Respir Cell MolBiol 51: 536-549.
- Santos LM, de BritoCervilha DA, Cabral LD, Garcia ÉK, Teixeira VP, et al. (2014) Bronchial responsiveness in an elastase-induced mouse model of emphysema. RespirPhysiolNeurobiol 194: 9-14.
- Onclinx C, De Maertelaer V, Gustin P, Gevenois PA(2006) Elastase-induced pulmonary emphysema in rats: comparison of computed density and microscopic morphometry. Radiology 241: 763-770.
- Furuya N, Takenaga M, Ohta Y, Tokura Y, Hamaguchi A, et al. (2012) Cell therapy with adipose tissue-derived stem/stromal cells for elastase-induced pulmonary emphysema in rats. Regen Med 7: 503-512.
- Bianchi A, Tibiletti M, Kjørstad Å, Birk G, et al. (2015) Three-dimensional accurate detection of lung emphysema in rats using ultra-short and zero echo time MRI. NMR Biomed 28: 1471-1479.
- Boiati RF, Manchini MT, Jacobsen O, Dos Santos Batista JG, Silva Júnior JA, et al. (2015) Evaluation of the Anti-inflammatory Activity of Atorvastatin and its Effect on Alveolar Diameter in a Model of Elastase-induced Emphysema in Rats. Drug Res (Stuttg) 65: 540-544.
- Szabari MV, Parameswaran H, Sato S, Hantos Z, Bartolák-Suki E, et al. (2012) Acute mechanical forces cause deterioration in lung structure and function in elastase-induced emphysema. Am JPhysiol Lung Cell MolPhysiol 303:567-574.
- Robertoni FS, Olivo CR, Lourenço JD, Gonçalves NG, Velosa AP, et al. (2015) Collagenase mRNA Overexpression and Decreased Extracellular Matrix Components Are Early Events in the Pathogenesis of Emphysema. PLoS One 10: e0129590.
- Takahashi A, Majumdar A, Parameswaran H, Bartolák-Suki E, Suki B (2014) Proteoglycans maintain lung stability in an elastase-treated mouse model of emphysema. Am J Respir Cell MolBiol 51: 26-33.
- Kuhn C, Yu SY, Chraplyvy M, Linder HE, Senior RM (1976) The induction of emphysema with elastase. II. Changes in connective tissue. Lab Invest 34: 372-380.
- Lucey EC, Goldstein RH, Stone PJ, Snider GL (1998) Remodeling of alveolar walls after elastase treatment of hamsters. Results of elastin and collagen mRNA in situ hybridization. Am J RespirCrit Care Med 158: 555-564.
- Ito S, Ingenito EP, Brewer KK, Black LD, Parameswaran H, et al. (2005) Mechanics, nonlinearity, and failure strength of lung tissue in a mouse model of emphysema: possible role of collagen remodeling. J ApplPhysiol 98: 503-511.
- Suki B, Ito S, Stamenovic D, Lutchen KR, Ingenito EP (2005) Biomechanics of the lung parenchyma: critical roles of collagen and mechanical forces. J ApplPhysiol 98: 1892-1899.
- Jesudason R, Sato S, Parameswaran H, Araujo AD, Majumdar A, et al. (2010) Mechanical forces regulate elastase activity and binding site availability in lung elastin. Biophys J 99: 3076-3083.
- Suki B, Jesudason R, Sato S, Parameswaran H, Araujo AD, et al. (2012) Mechanical failure, stress redistribution, elastase activity and binding site availability on elastin during the progression of emphysema. PulmPharmacolTher 25: 268-275.
- Limjunyawong N, Craig JM, Lagassé HA, Scott AL, Mitzner W (2015) Experimental progressive emphysema in BALB/cJ mice as a model for chronic alveolar destruction in humans. Am J Physiol Lung Cell MolPhysiol 309: L662-L676.
- Lüthje L, Raupach T, Michels H, Unsöld B, Hasenfuss G, et al. (2009) Exercise intolerance and systemic manifestations of pulmonary emphysema in a mouse model. Respir Res 10: 7.
- Kawakami M, Matsuo Y, Yoshiura K, Nagase T, Yamashita N (2008) Sequential and quantitative analysis of a murine model of elastase-induced emphysema. Biol Pharm Bull 31: 1434-1438.
- Lanzetti M, da Costa CA, Nesi RT, Barroso MV, Martins V, et al. (2012) Oxidative stress and nitrosative stress are involved in different stages of proteolytic pulmonary emphysema. Free RadicBiol Med 53: 1993-2001.
- Longhini-Dos-Santos N, Barbosa-de-Oliveira VA, Kozma RH, Faria CA, Stessuk T, et al. (2013) Cell therapy with bone marrow mononuclear cells in elastase-induced pulmonary emphysema. Stem Cell Rev 9: 210-218.
- Lopes FD, Toledo AC, Olivo CR, Prado CM, Leick EA, et al. (2013) A comparative study of extracellular matrix remodeling in two murine models of emphysema. HistolHistopathol 28: 269-276.
- Cruz FF, Antunes MA, Abreu SC, Fujisaki LC, Silva JD, et al. (2012) Protective effects of bone marrow mononuclear cell therapy on lung and heart in an elastase-induced emphysema model. RespirPhysiolNeurobiol 182: 26-36.
- Antunes MA, Abreu SC, Cruz FF, Teixeira AC, Lopes-Pacheco M, et al. (2014) Effects of different mesenchymal stromal cell sources and delivery routes in experimental emphysema. Respir Res 15: 118.
- Brusselle GG, Bracke KR, Maes T, D'hulst AI, Moerloose KB, et al. (2006) Murine models of COPD. PulmPharmacolTher 19: 155-165.
- D’Armiento J, Dalal SS, Okada Y, Berg RA, Chada K(1992) Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 71:955-961.
- Foronjy RF, Okada Y, Cole R, D'Armiento J (2003) Progressive adult-onset emphysema in transgenic mice expressing human MMP-1 in the lung. Am J Physiol Lung Cell MolPhysiol 284: L727-L737.
- Shiomi T, Okada Y, Foronjy R, Schiltz J, Jaenish R, et al. (2003) Emphysematous changes are caused by degradation of type III collagen in transgenic mice expressing MMP-1. Exp Lung Res 29: 1-15.
- Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, et al. (2000) Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med 192: 1587-1600.
- Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY (2000) Impaired distal airway development in mice lacking elastin. Am J Respir Cell MolBiol 23: 320-326.
- Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, et al. (2002) Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415: 171-175.
- Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, et al. (1996) PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85: 863-873.
- Lindahl P, Karlsson L, Hellström M, Gebre-Medhin S, Willetts K, et al. (1997) Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124: 3943-3953.
- Weinstein M, Xu X, Ohyama K, Deng CX (1998) FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125: 3615-3623.
- Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, et al. (2000) Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. ProcNatlAcadSci U S A 97: 5972-5977.
- Yoshida M, Korfhagen TR, Whitsett JA(2001) Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol 166: 7514-7519.
- Knudsen L, Wucherpfennig K, Mackay RM, Townsend P, Mühlfeld C, et al. (2009) A recombinant fragment of human surfactant protein D lacking the short collagen-like stalk fails to correct morphological alterations in lungs of SP-D deficient mice. Anat Rec (Hoboken) 292:183-189.
- Leco KJ, Waterhouse P, Sanchez OH, Gowing KL, Poole AR, et al. (2001) Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J Clin Invest 108: 817-829.
- Besnard V, Matsuzaki Y, Clark J, Xu Y, Wert SE, et al. (2010) Conditional deletion of Abca3 in alveolar type II cells alters surfactant homeostasis in newborn and adult mice. Am J Physiol Lung Cell MolPhysiol 298: L646-L659.
- Holm AT, Wulf-Johansson H, Hvidsten S, Jorgensen PT, Schlosser A, et al. (2015) Characterization of spontaneous air space enlargement in mice lacking microfibrillar-associated protein 4. Am J Physiol Lung Cell MolPhysiol 308: L1114-L1124.
- Martorana PA, van Even P, Gardi C, Lungarella G (1989) A 16-month study of the development of genetic emphysema in tight-skin mice. Am Rev Respir Dis 139: 226-232.
- Cavarra E, Bartalesi B, Lucattelli M, Fineschi S, Lunghi B, et al. (2001) Effects of cigarette smoke in mice with different levels of alpha(1)-proteinase inhibitor and sensitivity to oxidants. Am J RespirCrit Care Med 164: 886-890.
- Gardi C, Cavarra E, Calzoni P, Marcolongo P, de Santi M, et al. (1994) Neutrophil lysosomal dysfunctions in mutant C57Bl/6J mice: interstrain variations in content of lysosomalelastase, cathepsin G and their inhibitors. Biochem J 299: 237-245.
- O'Donnell MD, O'Connor CM, FitzGerald MX, Lungarella G, Cavarra E, et al. (1999) Ultrastructure of lung elastin and collagen in mouse models of spontaneous emphysema. Matrix Biol 18: 357-360.
- Groneberg DA, Chung KF (2004) Models of chronic obstructive pulmonary disease. Respir Res 5: 18.
- Mackay AJ, Hurst JR (2013) COPD exacerbations: causes, prevention, and treatment. Immunol Allergy Clin North Am 33: 95-115.
- Seemungal TA, Harper-Owen R, Bhowmik A, Jeffries DJ, Wedzicha JA (2000) Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. EurRespir J 16: 677-683.
- Stockley RA, O'Brien C, Pye A, Hill SL (2009) Relationship of sputum color to nature and outpatient management of acute exacerbations of COPD. 2000. Chest 136: e30.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, et al. (2005) Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 60: 925-931.
- Tanabe N, Muro S, Hirai T, Oguma T, Terada K, et al. (2011) Impact of exacerbations on emphysema progression in chronic obstructive pulmonary disease. Am J RespirCrit Care Med 183: 1653-1659.
- Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, et al. (2006) Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J RespirCrit Care Med 173: 1114-1121.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA (2002) Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 57: 847-852.
- Sethi S, Murphy TF (2008) Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 359: 2355-2365.
- Mehta H, Nazzal K, Sadikot RT (2008) Cigarette smoking and innate immunity. Inflamm Res 57: 497-503.
- Betsuyaku T1, Kuroki Y, Nagai K, Nasuhara Y, Nishimura M (2004) Effects of ageing and smoking on SP-A and SP-D levels in bronchoalveolar lavage fluid. EurRespir J 24: 964-970.
- Taylor DC, Cripps AW, Clancy RL (1995) A possible role for lysozyme in determining acute exacerbation in chronic bronchitis. ClinExpImmunol 102: 406-416.
- Gompertz S, Bayley DL, Hill SL, Stockley RA (2001) Relationship between airway inflammation and the frequency of exacerbations in patients with smoking related COPD. Thorax 56: 36-41.
- Berenson CS, Wrona CT, Grove LJ, Maloney J, Garlipp MA, et al. (2006) Impaired alveolar macrophage response to Haemophilus antigens in chronic obstructive lung disease. Am J RespirCrit Care Med 174: 31-40.
- Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S(2006) Impaired phagocytosis of nontypeableHaemophilus influenza by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis194:1375-1384.
- Rosell A, Monsó E, Soler N, Torres F, Angrill J, et al. (2005) Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med 165: 891-897.
- Sethi S, Maloney J, Grove L, Wrona C, Berenson CS (2006) Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J RespirCrit Care Med 173: 991-998.
- Fujimoto K, Yasuo M, Urushibata K, Hanaoka M, Koizumi T, et al. (2005) Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. EurRespir J 25: 640-646.
- Tsoumakidou M, Tzanakis N, Chrysofakis G, Siafakas NM (2005) Nitrosative stress, heme oxygenase-1 expression and airway inflammation during severe exacerbations of COPD. Chest 127: 1911-1918.
- Bathoorn E, Liesker JJ, Postma DS, Boorsma M, Bondesson E, et al. (2008) Anti-inflammatory effects of combined budesonide/formoterol in COPD exacerbations. COPD 5: 282-290.
- Bresser P, Out TA, van Alphen L, Jansen HM, Lutter R (2000) Airway inflammation in nonobstructive and obstructive chronic bronchitis with chronic Haemophilus influenza airway infection: comparison with noninfected patients with chronic obstructive pulmonary disease. Am J RespirCrit Care Med162: 947-952.
- Banerjee D, Khair OA, Honeybourne D (2004) Impact of sputum bacteria on airway inflammation and health status in clinical stable COPD. EurRespir J 23: 685-691.
- Marin A, Garcia-Aymerich J, Sauleda J, Belda J, Millares L, et al. (2012) Effect of bronchial colonization on airway and systemic inflammation in stable COPD. COPD9: 121-130.
- Crooks SW, Bayley DL, Hill SL, Stockley RA (2000) Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4. EurRespir J 15: 274-280.
- Aaron SD, Angel JB, Lunau M, Wright K, Fex C, et al. (2001) Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J RespirCrit Care Med 163: 349-355.
- Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders P,et al. (1996) Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J RespirCrit Care Med 154:813-816.
- Nowak D, Antczak A, Krol M, Pietras T, Shariati B, et al. (1996) Increased content of hydrogen peroxide in the expired breath of cigarette smokers. EurRespir J 9: 652-657.
- Woods JA, Wheeler JS, Finch CK, Pinner NA (2014) Corticosteroids in the treatment of acute exacerbations of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 9: 421-430.
- Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, et al. (2007) Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 356: 775-789.
- Calverley PM, Stockley RA, Seemungal TA, Hagan G, Willits LR, et al. (2011) Reported pneumonia in patients with COPD: findings from the INSPIRE study. Chest 139: 505-512.
- Sharafkhaneh A, Southard JG, Goldman M, Uryniak T, Martin UJ (2012) Effect of budesonide/formoterolpMDI on COPD exacerbations: a double-blind, randomized study. Respir Med 106: 257-268.
- Keatings VM, Jatakanon A, Worsdell YM, Barnes PJ (1997) Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am J RespirCrit Care Med 155: 542-548.
- Rahman I, Adcock IM (2006) Oxidative stress and redox regulation of lung inflammation in COPD. EurRespir J 28: 219-242.
- Ito K, Barnes PJ, Adcock IM(2000) Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12Mol Cell Biol 20: 6891-6903.
- Barnes PJ, Adcock IM (2009) Glucocorticoid resistance in inflammatory diseases. Lancet 373: 1905-1917.
- Quon BS, Gan WQ, Sin DD (2008) Contemporary management of acute exacerbations of COPD: a systematic review and metaanalysis. Chest 133: 756-766.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, et al. (2010) Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J RespirCrit Care Med181:150-157.
- Albert RK, Connett J, Bailey WC, Casaburi R, Cooper JA Jr, et al. (2011) Azithromycin for prevention of exacerbations of COPD. N Engl J Med 365: 689-698.
- Ram FS, Rodriguez-Roisin R, Granados-Navarrete A, Garcia-Aymerich J, Barnes NC. (2006) Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 19: CD004403.
- Mesaros N, Nordmann P, Plésiat P, Roussel-Delvallez M, Van Eldere J, et al. (2007) Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. ClinMicrobiol Infect 13: 560-578.
- Riou M, Carbonnelle S, Avrain L, Mesaros N, Pirnay JP, et al. (2010) In vivo development of antimicrobial resistance in Pseudomonas aeruginosa strains isolated from the lower respiratory tract of Intensive Care Unit patients with nosocomial pneumonia and receiving antipseudomonal therapy. Int J Antimicrob Agents 36:513-522.
- Stolk J, Rudolphus A, Davies P, Osinga D, Dijkman JH, et al. (1992) Induction of emphysema and bronchial mucus cell hyperplasia by intratracheal instillation of lipopolysaccharide in the hamster. J Pathol 167: 349-356.
- Kobayashi S, Fujinawa R, Ota F, Angata T, Ueno M, et al. (2013) A single dose of lipopolysaccharide into mice with emphysema mimics human chronic obstructive pulmonary disease exacerbation as assessed by micro-computed tomography. Am J Respir Cell MolBiol 49: 971-977.
- Spond J, Billah MM, Chapman RW, Egan RW, Hey JA, et al. (2004) The role of neutrophils in LPS-induced changes in pulmonary function in conscious rats. PulmPharmacolTher 17: 133-140.
- Hardaker EL, Freeman MS, Dale N, Bahra P, Raza F, et al. (2010) Exposing rodents to a combination of tobacco smoke and lipopolysaccharide results in an exaggerated inflammatory response in the lung. Br J Pharmacol160: 1985-1996.
- Nie YC, Wu H, Li PB, Luo YL, Zhang CC, et al. (2012) Characteristic comparison of three rat models induced by cigarette smoke or combined with LPS: to establish a suitable model for study of airway mucus hypersecretion in chronic obstructive pulmonary disease. PulmPharmacolTher 25: 349-356.
- Li Y, Gu C, Xu W, Yan J, Xia Y, et al. (2014) Therapeutic effects of amniotic fluid-derived mesenchymal stromal cells on lung injury in rats with emphysema. Respir Res 15: 120.
- Gaschler GJ, Skrtic M, Zavitz CC, Lindahl M, Onnervik PO, et al. (2009) Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J RespirCrit Care Med 179: 666-675.
- Huvenne W, Lanckacker EA, Krysko O, Bracke KR, Demoor T, et al. (2011) Exacerbation of cigarette smoke-induced pulmonary inflammation by Staphylococcus aureus enterotoxin B in mice. Respir Res 12: 69.
- Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, et al. (2004) Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J RespirCrit Care Med 170: 1164-1171.
- Voss M, Wonnenberg B, Honecker A, Kamyschnikow A, Herr C, et al. (2015) Cigarette smoke-promoted acquisition of bacterial pathogens in the upper respiratory tract leads to enhanced inflammation in mice. Respir Res 16: 41.
- Pang B, Hong W, West-Barnette SL, Kock ND, Swords WE (2008) Diminished ICAM-1 expression and impaired pulmonary clearance of nontypeableHaemophilusinfluenzae in a mouse model of chronic obstructive pulmonary disease/emphysema. Infect Immun 76:4959-4967.
- Wang D, Wang Y, Liu YN (2010) Experimental pulmonary infection and colonization of Haemophilusinfluenzae in emphysematous hamsters. PulmPharmacolTher 23: 292-299.
- Ganesan S, Faris AN, Comstock AT, Sonstein J, Curtis JL, et al. (2012) Elastase/LPS-exposed mice exhibit impaired innate immune responses to bacterial challenge: role of scavenger receptor A. Am J Pathol 180: 61-72.
- Bauer CM, Zavitz CC, Botelho FM, Lambert KN, Brown EG, et al. (2010) Treating viral exacerbations of chronic obstructive pulmonary disease: insights from a mouse model of cigarette smoke and H1N1 influenza infection. PLoS One 5: e13251.
- Singanayagam A, Glanville N, Walton RP, Aniscenko J, Pearson RM, et al. (2015) A short-term mouse model that reproduces the immunopathological features of rhinovirus-induced exacerbation of COPD. ClinSci (Lond) 129:245-258.