Keywords
Colorectal cancer; Fisetin; Endopeptidases; Bioavailability; Inhibition
Abbreviations
CRC: Colorectal Cancer; AC: Classical Adenocarcinomas; MAC: Mucinous Adenocarcinoma; SC: Signet-ring Cell Carcinomas; EMT: Epithelial-Mesenchymal Transition; MMPs: Matrix Metalloproteinases; MMP-8: Matrix Metalloproteinase 8; MMP-13: Matrix Metalloproteinase 13; VEGF: Vascular Endothelial Growth Factors; LD50: Lethal Dose 50; ADME: Absorption, Distribution, Metabolism, Excretion; 5XT and 1UA: Co-crystallised Inhibitors; CYP: Cytochrome P450.
Introduction
Colorectal cancer (CRC) is third on the list of the most prevalently diagnosed malignancies and the fourth major cause of cancer-related death worldwide [1-4]. CRC incidence is forecast to increase by about 60% to more than 2.2 million new cases and almost 1.1 million deaths by 2030 [5]. Even with the remarkable headways in diagnosis, screening and treatment of CRC, the comprehensive long-term outcome of curatively resected patients have not noticeably changed in the last few decades, the 5-year survival rate being approximately 60%, primarily dependent on the stage at diagnosis [6,7]. Histological subtypes according to World Health Organization (WHO) were defined as classical adenocarcinomas (AC), accounting for the majority of cases. The mucinous adenocarcinomas (MAC), signet-ring cell carcinomas (SC), and other even less occurring forms (small cell carcinoma, squamous cell carcinoma, adenosquamous carcinoma, medullary carcinoma, and undifferentiated carcinoma) also exist [6,8]. Colorectal neoplasm develops slowly, over years or even decades. Consequently, most CRC commence with the polyps occurring on the epithelial lining of the colon or rectum. These abnormal growths may be hyperplastic polyp (benign), tubular adenoma (pre-malignant) or colorectal adenocarcinoma (malignant) [9]. Although it is calculated that about 20% occurrences of CRC have familial history of the tumour and some genetic syndromes accounts for about 4% of people with greater risks of CRC [9]. However, most of the CRC cases are connected to environmental factors rather than genetic alterations. Incidences of CRC may depend on numerous factors such as diets, life style, intestinal microbiota, mutagens and other environmental parameters [5,9-11]. Many of the CRC patients in Dukes A and B (early) stage can be treated successfully by surgical resection. Nevertheless, the malignant CRC that undergoes metastasis in the advanced stage (Dukes C and D) is conventionally refractory to available therapies and carry quite a poor prognosis, leading to the consequential drop in the 5-year survival rates [10,12,13]. During progression to metastasis, a subset or individual cancer cells undergo epithelial-mesenchymal transition (EMT), detach from the primary tumour and migrate to the blood or lymph, colonizing distant organs or tissues such as liver, lung and peritoneum. In addition, various other metastatic sites are the spleen, bone, the brain and rarely, the pancreas and heart [9,12,14].
Epithelial-mesenchymal transition (EMT) is a vital regulator of tissues and organ formation, which is implicated as requisite mechanism in cancer progression and metastasis [15]. EMT allows epithelium-derived tumour cells metastasis by promoting loss of cell adhesion and gain of migratory together with invasive properties [16,17]. The reduction in E-cadherin expression is a crucial factor in EMT process. The single-span transmembrane glycoprotein E-cadherin interacts with adjacent E-cadherin molecules expressed by proximate cells and moderates calcium-dependent intercellular adhesion [18]. The loss or reduction in E-cadherin expression is prompted by chromosomal deletions, DNA methylation, proteolytic cleavage by matrix metalloproteinases (MMPs) [19] and several transcriptional repressors such as Snail, Slug and Twist1 as well as other direct and indirect repressors [20,21]. Previous studies have depicted that abnormal E-cadherin expression is linked to high-grade and poorly differentiated tumour stage for numerous types of cancers including breast cancer, lung cancer, gastric cancer, prostate cancer and colorectal cancer [21].
Fundamental cancer studies have primarily focused on either gainof- function in oncogenes or loss-of-function in the tumour suppressor genes [22], which are sequel to the genetic alterations that characterise tumour cells [23-25]. However, the extracellular matrix of the cancer cells and the stromal cells within the tumour microenvironment possess a significant effect on tumour progression [23,24]. Matrix metalloproteinases (MMPs; also known as matrixins), family extracellular zinc-dependent endopeptidases [26], are involved in the degradation and remodelling of extracellular matrix in physiological and pathological processes [27]. MMPs also have a crucial role in cell proliferation, differentiation, migration, angiogenesis, immune responses and apoptosis, which are implicated in the hallmarks of CRC [7,12,13,28].
MMP-8 has been considered as a double-edged sword due to its equivocal role in cancer progression [29]. Previous studies showed that MMP-8 might play some beneficial roles during tumour progression [15,30]. For instance, the loss of MMP-8 significantly increases the incidence of skin cancer in male MMP-8-/- mice and MMP-8-deficient female mice develop tongue cancer more often than wild-type mice [3,31]. However, other reports noted that the up-regulation of MMP- 8 is directly proportional to the progression of colorectal cancer [29]. In addition, there is a positive correlation between MMP-8 and TGF-β1, which confers the acquired invasion and metastasis through the induction of EMT [29]. MMP-13, also named collagenase-3 [32], is another MMP, implicated in the remodelling of the ECM components important to cancer microenvironments. Under pathological conditions such as in CRC, MMP-13 expression is up-regulated by cancer invasion-related factors [33] and other studies suggest that MMP-13 may play a central role in the extracellular MMP activation cascade in cancer microenvironment [34]. Although high expression of MMP-13 is directly linked to the invasiveness and poor prognosis of CRC [35], previous findings (ref) has also established that the enzyme is involved in tumour angiogenesis [36]. MMP-13 elevated in the tumour microenvironments promotes angiogenesis through increased secretions of vascular endothelial growth factors (VEGF) in cancer invasive area [33,37]. Therefore, the regulation of the activities of MMP-8 and MMP-13 is important in the treatment of CRC.
Therapeutic inhibitors of MMPs (MMPI) have been developed, but clinical trials for these drugs as regards to cancer treatments have proved disappointing [38]. The failures can be explained based on the followings: some of the problems are associated with the MMPI themselves, such as poor oral bioavailability, broad spectrum inhibition [7], metabolic instability and dose-limiting toxicity, while others depend on the flops linked with the clinical trials such as poor trial design and the use of inadequate clinical end-points [24,26]. With the apt hindsight and the understanding of the diverse role of MMPs and their relevance in the early progression of cancer diseases [39], such as angiogenesis — we could improve on these disappointing results. To better target MMPs for cancer treatment, we must thoroughly consider the specific cancer-types and stages of disease and the expression profile together with the activity of MMPs in the different stages of the tumour progression [31].
Fisetin (3,7,3,4-tetrahydroxyflavone) is a flavonoid, commonly found in plants such as Annona muricata, Acacia greggii, Butea frondosa, Acacia berlandieri, Gleditsia triacanthos and Quebracho coloradocs [40]. It is also widely present in fruits and vegetables such as persimmon, strawberry, apple, grape, onion and cucumber at concentrations of 2-160 mg/g [41] (Figure 1).
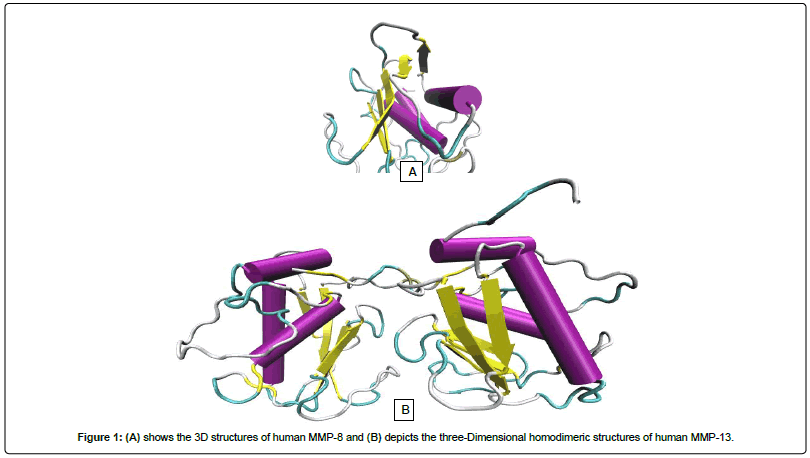
Figure 1: (A) shows the 3D structures of human MMP-8 and (B) depicts the three-Dimensional homodimeric structures of human MMP-13.
Fisetin has been reported to possess anti-inflammatory [42], antioxidant [43], anticancer [40], antihyperlipidaemic, and neuroprotective properties [44], which can be explored for more therapeutic purposes. Fisetin has been shown to target many components of intracellular signalling pathways including controlled mechanisms of cell survival and apoptosis, tumour angiogenic and metastatic switches by modulating a peculiar set of upstream kinases, transcription factors and their regulators. Patently, there are enormous evidence which buttresses the idea that fisetin is a promising therapeutics for CRC treatment [40,45]. In this study, Insilico approach was employed to examine the inhibitory potentials of fisetin against catalytically important amino acid residues in the S1 sites of the targets MMP-8 and MM-13. The flavonoid's ADMET (Absorption, Distribution, Metabolism and Excretion) showed that fisetin obeyed Lipinski's rule of five [46] and the toxicity results showed that the compound belongs to toxicity class 3 with LD50 of 159 mg/ kg. Consequently, the ADMET and bioavailability properties of fisetin portray it as a probable drug-like candidate with satisfactory oral bioavailability. Nevertheless, more work needs to be done to elucidate the role of fisetin on the some members of the cytocrome P450 family.
Materials and Methods
Preparation of ligands
The 2D chemical conformation of Fisetin was downloaded from PubChem, a popular database for the retrieval of ligands. The 3D structure of Fisetin was created using Babel 2.3.2, and Autodock MGLTool-1.5.6 was used to produce the pdbqt format of ligands.
Preparation of enzymes
X-ray crystal structures of the human enzymes were retrieved from RCSB (Research Collaboratory for Structural Bioinformatics) Protein Data Bank. The PDB ID of the enzymes are 5H8X (MMP-8) and 4L19 (MMP-13). All water molecules were removed and the pdqt file for each protein was generated using the molecular graphic program PyMOL. Config files was generated, and the grid coordinates was exactly as that of the co-crystallized compounds in order to ensure that Fisetin occupies the same binding pocket as obtained from MMP-8 (5H8X, 1.3 Å, x=-9.65, y=-10.32, z=20.29) and MMP-13 (4L19 1.66 Å, x=14.4 y=16.3, z=24.51).
Molecular docking
Molecular docking was carried out using AutoDock4.2. Command lines were used to dock the ligand in the same binding pockets in each enzyme targets occupied by their co-crystalized compounds.
ADMET and bioavailability scores
The ADME (absorption, distribution, metabolism and excretion) properties for Fisetin were calculated by using smile notation in Swiss ADME web based tool [47]. Furthermore, the toxicity properties like LD50 for fisetin was calculated by using PROTOX web based tool [48]. The bioavailability scores were calculated by using Molinspiration web based tool [49].
Enzyme-ligand complex comformational analysis
We complexed the ligands (fisetin or the cocrystalised 5XT and 1UA) and each enzyme separately in the pdb format using PyMol and then submitted it on ProteinsPlus, an online server [50]. ProteinsPlus automatically generated the PoseView (2D) diagrams of the macromolecular complexes. In addition, another online server PLIP was applied to depict the 3-Dimensional conformation of each complex.
Results and Discussion
Molecular docking
Fisetin showed a better binding affinity for the target enzyme than the cocrystalised 5XT for MMP-8 and 1UA in MMP-13 has indicated by Table 1: Fisetin is present in many fruits and vegetables. Preclinical studies on fisetin has shown potential inhibition of cancer growth through alteration of cell cycle, prompts apoptosis, angiogenesis, invasion, and metastasis without inducing significant adverse events to normal cells [40,45]. Previous studies had focused majorly on the ability of fisetin to targets many components of intracellular signalling pathways which involve regulators of cell survival and apoptosis, tumour angiogenic and metastatic switches by regulating the different set of upstream kinases, transcription factors and their regulators [40,51]. Molecular docking studies are important to predict the most stable orientation of one molecule to the other when complexed together. In this study, we explored the therapeutic inhibitory effect of fisetin on the catalytic sites of MMP-8 and MMP- 13, which are both collagenases and its effects on colorectal cancer together with other tumours. We analysed the structural interactions of fisetin against catalytic important amino acid residues in the S1 sites of the targets. The results of the molecular docking of MMP-8 with fisetin gave significant binding energies of -10.0 kcal/mol. The docking score of MMP-13-fisetin complex was -9.9 kcal/mol. From this result, it is obvious that fisetin showed higher binding affinity for the enzyme targets than the cocrystalised inhibitors.
| Enzyme Targets |
Binding Energy for 5XT and 1UA (kcal/mol) |
Binding Energy for Fisetin (kcal/mol) |
| MMP-8 |
-9.9 |
-10.0 |
| MMP-13 |
-9.3 |
-9.9 |
Table 1: Docking Results for Co-crystallised Inhibitor vs Fisetin
Post docking analysis
6.2.1 Fisetin partially distort the S1 sites while partly maintaining the hydrophobic pockets: Up next, we carefully resolved the order of interacting bond pairs in the light of previously described MMPinhibitors such as 5XT and 1UA [52,53]. We predicted the chemical interactions of our putative ligand/protein complex, not just as a validation procedure, but also to provide insights into the efficacy of receptor binding and propensity to complex dissociation. 5XT, a catechol-containing chemical entity which is a well described MMP- 8 inhibitor was first resolved and benchmarked for studying the test compounds. Apart from situating itself in a deep and proximate orientation within MMP-8’s binding pocket (S1), a highly stable zigzagged network of hydrogen bond within ~3.0 Å bond distance was observed in Figures 2-4, between the amine ports of the backbone residues Leu160 and Ala161 and sulfonamide moiety of the standard inhibitor [52]. These residues have been reported to be crucial in MMP-8 inhibition. An extra benzene ring (R3) of 5XT, which harbours two OH- groups efficiently, twists to optimally engage in a hydrogen trade-off with catalytic important Ala163, thus disrupting the pocket’s orientation. Our observations about the hydrogen network outside the hydrophobic shell are in consonance with a recent report [52] where little information was provided about the lower hydrophobic shell. In exploring this, we found that rings 1 and 2 protrude deeply well within an extensive hydrophobic pocket spanning Leu193 to Tyr219 (Figure 3B). The current catechol-based MMP-8 inhibitor pose is crucial for impeding pocket hydration and ligand desolvation over time, and as such, we conclude that steric, electrostatic and hydrophobic interactions complement each other for efficiently antagonizing MMP-8.
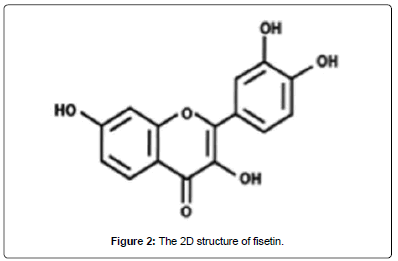
Figure 2: The 2D structure of fisetin.
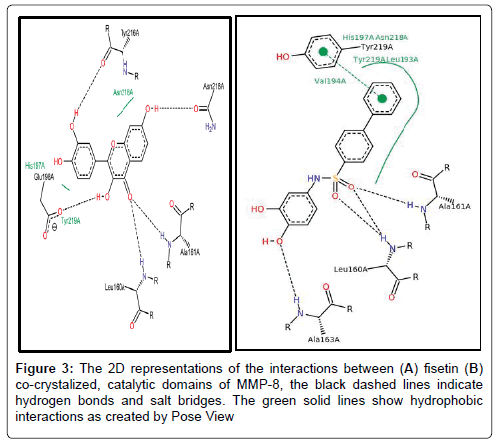
Figure 3: The 2D representations of the interactions between (A) fisetin (B) co-crystalized, catalytic domains of MMP-8, the black dashed lines indicate hydrogen bonds and salt bridges. The green solid lines show hydrophobic interactions as created by Pose View
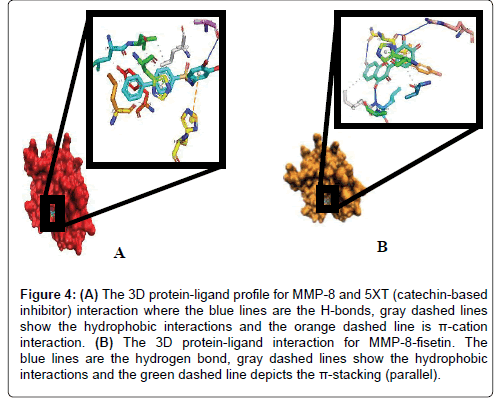
Figure 4: (A) The 3D protein-ligand profile for MMP-8 and 5XT (catechin-based inhibitor) interaction where the blue lines are the H-bonds, gray dashed lines show the hydrophobic interactions and the orange dashed line is π-cation interaction. (B) The 3D protein-ligand interaction for MMP-8-fisetin. The blue lines are the hydrogen bond, gray dashed lines show the hydrophobic interactions and the green dashed line depicts the π-stacking (parallel).
To our surprise, when we explored the bonding network proposed by fisetin (a flavonol-mimetic), Leu160, Ala161, Asn218, Tyr 219 and Arg 222 were efficiently engaged within an impressive ~3.0 Å hydrogen bonding distance. The carbonyl oxygen of fisetin formed H-bond networks with the parallel LEU160 and ALA161 as compared to previous studies. However, hydrophobic patches between His 197 to Asn 218 through to Tyr 219 were scattered round the ligand as shown by Figures 3B and 4A. The prominent interaction in the S1 pocket as established by previous researchers (ref) is ideally the planer stacking of phenyl ring and HIS197. Upon visual inspection with VMD, the previously hollow S1 looks distorted and shredded, thereby enabling access to water molecules. This is also evident in the patched hydrophobes surrounding fisetin. We could not infer any reason for this since the docking algorithm accounts for a rigid protein. Howbeit, it is probable that the VMD algorithm could best represent the fisetin/ MMP-8 complex this way. Another astonishing fact is that fisetin fits well to previously described QSAR criteria as per hydrophobicity and electrostatic interactions. We insinuate that apart from antagonizing MMP-8, fisetin’s orientation further alters the hollow coordination within the comparatively restricted S1, thus efficiently incapacitating MMP-8 in CRC progression.
We also studied the binding modes of a hypothetical MMP-13/ fisetin complex. Prior to this we analysed the chemical behaviour of the exosite-binding 1UA inhibitor with MMP-13’s crucial residues as it has been previously reported. Noteworthy, is the protrusion of the P-methylphenyl moiety of 1UA into the hydrophobic S1 pocket as indicated in Figures 5 and 6, pointing towards the substrate binding cleft and contacted by the hydrophobic region, which contains residues, TYR244, VAL219, HIS222, Leu239 [53]. In addition, there was a planar stacking interaction between the imidazole ring of HIS222 and the compound 1UA phenyl ring. Aside these, ligand derived N19 is protonated by the carbonyl portion of the crucial Thr245 thus forming a hydrogen bond (Figure 5). Again, we report a similar network for fisetin with disconnected but extensive hydrophobic patches, where the residues LEU185, VAL219, TYR244 and HIS222 form a scattered hydrophobic network around the fisetin molecule in Figures 5 and 6. Furthermore, a similar π-π stacking interaction was observed the imidazole ring of HIS222 and a phenyl moiety of fisetin (Figure 6), as compared to the co-crystallized 1UA inhibitor (Figure 6) [53]. Fisetin partially twists in a g-conformation within the hollow S1 thus enabling optimal water-assisted hydrogen-bond contacts with GLU223, ALA186, LEU185, LEU239 just outside the hydrophobic shell while maintaining the necessary hydrophobic bonds.
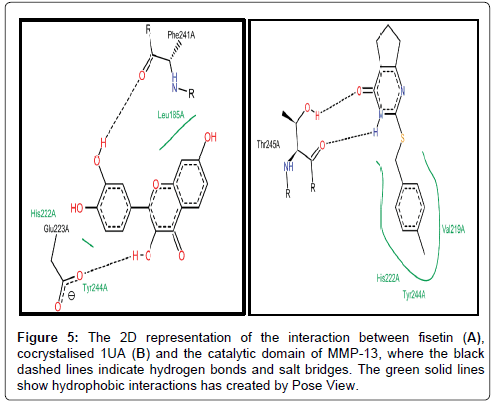
Figure 5: The 2D representation of the interaction between fisetin (A), cocrystalised 1UA (B) and the catalytic domain of MMP-13, where the black dashed lines indicate hydrogen bonds and salt bridges. The green solid lines show hydrophobic interactions has created by Pose View.
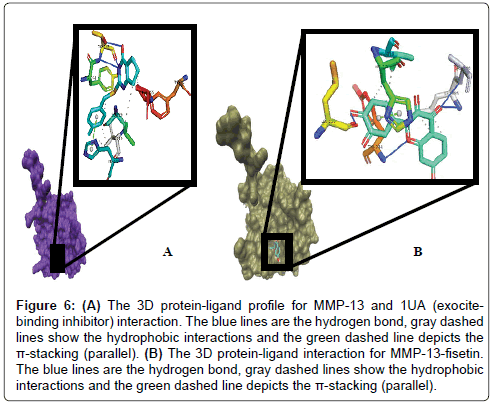
Figure 6: (A) The 3D protein-ligand profile for MMP-13 and 1UA (exocitebinding inhibitor) interaction. The blue lines are the hydrogen bond, gray dashed lines show the hydrophobic interactions and the green dashed line depicts the π-stacking (parallel). (B) The 3D protein-ligand interaction for MMP-13-fisetin. The blue lines are the hydrogen bond, gray dashed lines show the hydrophobic interactions and the green dashed line depicts the π-stacking (parallel).
Overall, in both cases (MMP-8/-13), we noted (and justified by previous studies, [52,53]) that ideally, previously described inhibitors aggregate require a decisive combo of hydrophobic and hydrogen bonds for antagonism. These were consistent with the binding signatures presented by fisetin.
MMPs have been elucidated to play significant role in the development and progression of CRC [29,33,37]. Previous reports [3,29,54] showed that MMP-8 may have some protective activities, thereby discouraging its inhibition in cancer treatment. Howbeit, other studies indicate that up-regulation of MMP-8 is positively correlated with the progression of CRC [3,29,54]. Therefore, the timely inhibition of MMP-8 is justified. MMP-13 activity is importantly higher in the tumour tissues than normal colonic mucosa, where it acts as an indispensable mediator of ECM degradation implicated in CSC tumour angiogenesis, invasion and metastasis [33-35]. In order to circumvent the shortcomings of previous clinical trials for MMP inhibitors, we must thoroughly consider the specific cancer-types and stages of disease and the expression profile together with the activity of MMPs in the different stages of the tumour progression [24]. In this study, fisetin satisfactorily inhibits the post-translational forms of MMP-8 and -13, thereby suppressing ECM modifications necessary for tumour angiogenesis, invasion and metastasis.
ADMET properties
Fisetin was found to follow Lipinski's rule of five: ADMET properties and bioactivity scores of fisetin are depicted in Tables 2-8. The lead compound was shown to follow Lipinski’s rule of 5 which underscore fisetin as a potent drug as observed in Tables 2 and 6. The toxicity results showed that the compound belongs to toxicity class 3 with LD50 of 159 mg/kg. This provided evidence for its least toxicity or no toxicity if administered at a dosage far less than 100 mg/kg.
| Formula |
C15H10O6 |
| Molecular weight |
286.24 g/mol |
| Num. heavy atoms |
21 |
| Num. arom. heavy atoms |
16 |
| Fraction Csp3 |
0.00 |
| Num. rotatable bonds |
1 |
| Num. H-bond acceptors |
6 |
| Num. H-bond donors |
4 |
| Molar Refractivity |
76.01 |
| TPSA |
111.13 Ų |
Table 2: Physicochemical Properties of Fisetin.
| Log Po/w (iLOGP) |
1.50 |
| Log Po/w (XLOGP3) |
1.97 |
| Log Po/w (WLOGP) |
2.28 |
| Log Po/w (MLOGP) |
-0.03 |
| Log Po/w (SILICOS-IT) |
2.03 |
| Consensus Log Po/w |
1.55 |
Table 3: Lipophilicity of Fisetin.
| Log S (ESOL) |
-3.35 |
| Solubility |
1.27e-01 mg/ml; 4.43e-04 mol/l |
| Class |
Soluble |
| Log S (Ali) |
-3.93 |
| Solubility |
3.37e-02 mg/ml; 1.18e-04 mol/l |
| Class |
Soluble |
| Log S (SILICOS-IT) |
-3.82 |
| Solubility |
4.29e-02 mg/ml; 1.50e-04 mol/l |
| Class |
Soluble |
Table 4: Water Solubility of Fisetin.
| GI absorption |
High |
| BBB permeant |
No |
| P-gp substrate |
No |
| CYP1A2 inhibitor |
Yes |
| CYP2C19 inhibitor |
No |
| CYP2C9 inhibitor |
No |
| CYP2D6 inhibitor |
Yes |
| CYP3A4 inhibitor |
Yes |
| Log Kp (skin permeation |
-6.65 cm/s |
Table 5: Pharmacokinetics of Fisetin.
| Lipinski |
Yes; 0 violation |
| Ghose |
Yes |
| Veber |
Yes |
| Egan |
Yes |
| Muegge |
Yes |
| Bioavailability Score |
0.55 |
Table 6: Druglikeness of Fisetin.
| PAINS |
1 alert: catechol_A |
| Brenk |
1 alert: catechol |
| Leadlikeness |
Yes |
| Synthetic accessibility |
3.16 |
Table 7: Medicinal Chemistry of Fisetin.
| GPCR ligand |
-0.11 |
| Ion channel modulator |
-0.27 |
| Kinase inhibitor |
0.18 |
| Nuclear receptor ligand |
0.20 |
| Protease inhibitor |
-0.36 |
| Enzyme inhibitor |
0.20 |
Table 8: Molinspiration bioactivity score of Fisetin.
Susceptibility of the human body to carcinogens has noticeably increased in the last few decades. People are exposed to xenobiotics from environmental pollutants, food contaminants, pesticides, and drugs [55]. Therefore, the cytochrome P450 enzymes involved in phase I drug metabolism either detoxify or biotransform these foreign agents, leading to carcinogenesis. In vitro, flavonoids such as fisetin predominantly inhibit the major phase I drug-metabolizing enzyme CYP450 3A4 and the enzymes responsible for the bioactivation of procarcinogens (CYP1 enzymes) and up-regulate the enzymes involved in carcinogen detoxification (UDP-glucuronosyltransferases and glutathione S-transferases) [55,56]. The inhibition of CYP1A2, a procarcinogen-bioactivating enzyme as indicated in Table 5 may underscore the chemoprotective effect of fisetin as reported in previous studies [40,44]. Furthermore, CYP3A4 is a major cytochrome P450 enzyme, coupled with its abundant expression in liver; CYP3A4 is involved in metabolizing many anticancer agents [55]. The effect of fisetin on CYP3A4 as depicted by Table 5 may noticeably influence the bioactivity and bioavailability of enterally-administered anticancer drugs which are CYP3A4 substrate [55]. Obvious, this shows that fisetin can be co-administered with other potent antitumour drugs to synergistically leading to CRC remission. Since this studies partly corroborate with the previous findings elucidating the relationship between flavonoids and CYP [55,56], the effect of fisetin on phase I drug-metabolizing enzymes are ambiguous and difficult to predict. Therefore, further studies are essential to elucidate the grey areas associated with the modulatory activities of fisetin on CYP enzymes.
Conclusion
Unequivocally, this in-silico study provides evidence that the inhibitory potential of fisetin on MMP-8 and -13 is connected to distortion of the catalytic sites of these proteases, while partly interacting with some hydrophobic residues. These results confirm previous studies, which suggest fisetin as a potent anticancer agent, not just powerful to arrest key cell cycle modulators implicated in tumour progression, but it can also inhibit post-translational MMPs responsible for ECM remodelling. In addition, the ADMET and bioavailability properties of fisetin portray it as a probable drug-like candidate with satisfactory oral bioavailability. Nevertheless, more work needs to be done to elucidate the roles of fisetin on some members of the cytochrome P450 family.
Acknowledgements
We acknowledge the training and technical support received from researchers at the Centre for Biocomputing and Drug Development, Adekunle Ajasin University, Akungba-Akoko, Ondo State, Nigeria.
21812
References
- Ligi D, Mannello F (2016) Do matrix metalloproteinases represent reliable circulating biomarkers in colorectal cancer? Nature Publishing Group.
- Siminoff LA, Rogers HL, Harris-Haywood S (2015) Missed opportunities for the diagnosis of colorectal cancer. BioMed Research International.
- Väyrynen JP, Vornanen J, Tervahartiala T, Sorsa T, Bloigu R, et al. (2012) Serum mmp8 levels increase in colorectal cancer and correlate with disease course and inflammatory properties of primary tumors. International Journal of Cancer 131.
- Williams MD, Xian L, Huso T, Park JJ, Huso D, et al. (2016) Fecal metabolome in hmga1 transgenic mice with polyposis: Evidence for potential screen for early detection of precursor lesions in colorectal cancer. Journal of Proteome Research 15: 4176-4187.
- Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, et al. (2016) Global patterns and trends in colorectal cancer incidence and mortality. Gut J 2015: 310912.
- Hugen N, Van de Velde C, De Wilt J, Nagtegaal I (2014) Metastatic pattern in colorectal cancer is strongly influenced by histological subtype. Annals of Oncology 25: 651-657.
- Said AH, Raufman JP, Xie G (2014) The role of matrix metalloproteinases in colorectal cancer. Cancers 6: 366-375.
- Nitsche U, Zimmermann A, Späth C, Müller T, Maak M (2013) Mucinous and signet-ring cell colorectal cancers differ from classical adenocarcinomas in tumor biology and prognosis. Annals of Surgery 258: 775.
- Bardhan K, Liu K (2013) Epigenetics and colorectal cancer pathogenesis. Cancers 5: 676-713.
- Herszényi L, Hritz I, Lakatos G, Varga MZ, Tulassay Z (2012) The behavior of matrix metalloproteinases and their inhibitors in colorectal cancer. International Journal of Molecular Sciences 13: 13240-13263.
- Thanki K, Edward Nicholls M, Gomez G, Gajjar A, James Senagore A, et al. (2017) Consensus molecular subtypes of colorectal cancer and their clinical implications. International Biological and Biomedical Journal 3: 105-111.
- Mook OR, Frederiks WM, Van Noorden CJ (2004) The role of gelatinases in colorectal cancer progression and metastasis. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 1705: 69-89.
- Zucker S, Vacirca J (2004) Role of matrix metalloproteinases (mmps) in colorectal cancer. Cancer and Metastasis Reviews 23: 101-117.
- Yang B, Gao J, Rao Z, Shen Q (2012) Clinicopathological significance and prognostic value of mmp-13 expression in colorectal cancer. Scandinavian Journal of Clinical and Laboratory Investigation 72: 501-505.
- Ala-aho R, Kähäri VM (2005) Collagenases in cancer. Biochimie 87: 273-286.
- Garg M (2017) Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Reviews in Molecular Medicine, p: 19.
- Tsai JH, Yang J (2013) Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes & Development 27: 2192-2206.
- Stoops SL, Pearson AS, Weaver C, Waterson AG, Days E, et al. (2011) Identification and optimization of small molecules that restore e-cadherin expression and reduce invasion in colorectal carcinoma cells. ACS Chemical Biology 6: 452-465.
- Chabottaux V, Noel A (2007) Breast cancer progression: Insights into multifaceted matrix metalloproteinases. Clinical & Experimental Metastasis 24: 647-656.
- Palmer MB (2008) Regulation of snail transcription in cancer. Emory University.
- Wang JR, Gan WJ, Li XM, Zhao YY, Li Y, et al. (2014) Orphan nuclear receptor nur77 promotes colorectal cancer invasion and metastasis by regulating mmp-9 and e-cadherin. Carcinogenesis 35: 2474-2484.
- Brown GT, Murray GI (2015) Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. The Journal of Pathology 237: 273-281.
- Cathcart J, Pulkoski-Gross A, Cao J (2015) Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes & Diseases 2: 26-34.
- Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nature Reviews Cancer 2: 161-174.
- Ishii G, Ochiai A, Neri S (2016) Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Advanced Drug Delivery Reviews 99: 186-196.
- Vandenbroucke RE, Libert C (2014) Is there new hope for therapeutic matrix metalloproteinase inhibition? Nature Reviews Drug Discovery 13: 904-927.
- Yadav L, Puri N, Rastogi V, Satpute P, Ahmad R, et al. (2014) Matrix metalloproteinases and cancer-roles in threat and therapy. Asian Pac J Cancer Prev 15: 1085-1091.
- Crotti S, Piccoli M, Rizzolio F, Giordano A, Nitti D, et al. (2017) Extracellular matrix and colorectal cancer: How surrounding microenvironment affects cancer cell behavior? Journal of Cellular Physiology 232: 967-975.
- Qin G, Luo M, Chen J, Dang Y, Chen G, et al. (2016) Reciprocal activation between mmp-8 and tgf-β1 stimulates emt and malignant progression of hepatocellular carcinoma. Cancer Letters 374: 85-95.
- Radisky ES, Sarmazdeh MR, Radisky DC (2017) Therapeutic potential of matrix metalloproteinase inhibition in breast cancer. Journal of Cellular Biochemistry.
- Gialeli C, Theocharis AD, Karamanos NK (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. The FEBS Journal 278: 16-27.
- Gong Y, Chippada Venkata UD, Oh WK (2014) Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 6: 1298-1327.
- Kudo Y, Iizuka S, Yoshida M, Tsunematsu T, Kondo T, et al. (2012) Matrix metalloproteinase-13 (mmp-13) directly and indirectly promotes tumor angiogenesis. Journal of Biological Chemistry 287: 38716-38728.
- Leeman M, McKay J, Murray GI (2002) Matrix metalloproteinase 13 activity is associated with poor prognosis in colorectal cancer. Journal of Clinical Pathology 55: 758-762.
- Yamada T, Oshima T, Yoshihara K, Tamura S, Kanazawa A, et al. (2010) Overexpression of mmp-13 gene in colorectal cancer with liver metastasis. Anticancer Research 30: 2693-2699.
- Li Y, Sun B, Zhao X, Wang X, Zhang D, et al. (2017) Mmpâ€ÂÂÂÃÂ2 and mmpâ€ÂÂÂÃÂ13 affect vasculogenic mimicry formation in large cell lung cancer. Journal of Cellular and Molecular Medicine.
- Cirri P, Chiarugi P (2012) Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer and Metastasis Reviews 31: 195-208.
- Atay S, Banskota S, Crow J, Sethi G, Rink L, et al. (2014) Oncogenic kit-containing exosomes increase gastrointestinal stromal tumor cell invasion. Proceedings of the National Academy of Sciences 111: 711-716.
- Hadler-Olsen E, Winberg JO, Uhlin-Hansen L (2013) Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumor Biology 34: 2041-2051.
- Rengarajan T, Yaacob NS (2016) The flavonoid fisetin as an anticancer agent targeting the growth signaling pathways. European Journal of Pharmacology 789: 8-16.
- Koneru M, Sahu BD, Kumar JM, Kuncha M, Kadari A, et al. (2016) Fisetin protects liver from binge alcohol-induced toxicity by mechanisms including inhibition of matrix metalloproteinases (mmps) and oxidative stress. Journal of Functional Foods 22: 588-601.
- Chien CS, Shen KH, Huang JS, Ko SC, Shih YW (2010) Antimetastatic potential of fisetin involves inactivation of the pi3k/akt and jnk signaling pathways with downregulation of mmp-2/9 expressions in prostate cancer pc-3 cells. Molecular and Cellular Biochemistry 333: 169.
- Khan N, Syed DN, Ahmad N, Mukhtar H (2013) Fisetin: A dietary antioxidant for health promotion. Antioxidants & Redox Signalling 19: 151-162.
- Akaishi T, Morimoto T, Shibao M, Watanabe S, Sakai-Kato K, et al. (2008) Structural requirements for the flavonoid fisetin in inhibiting fibril formation of amyloid β protein. Neuroscience Letters 444: 280-285.
- Leu JD, Wang BS, Chiu SJ, Chan CY, Chen CC, et al. (2016) Combining fisetin and ionizing radiation suppresses the growth of mammalian colorectal cancers in xenograft tumor models. Oncology Letters 12: 4975-4982.
- Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug discovery today. Technologies 1: 337-341.
- Daina A, Michielin O, Zoete V (2017) Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 7: 42717.
- Drwal MN, Banerjee P, Dunkel M, Wettig MR, Preissner R (2014) Protox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Research 42: W53-W58.
- Jeliazkova N (2012) Web tools for predictive toxicology model building. Expert Opinion on Drug Metabolism & Toxicology 8: 791-801.
- Fährrolfes R, Bietz S, Flachsenberg F, Meyder A, Nittinger E, et al. (2017) Proteinsplus: A web portal for structure analysis of macromolecules. Nucleic Acids Research.
- Suh Y, Afaq F, Khan N, Johnson JJ, Khusro FH, et al. (2010) Fisetin induces autophagic cell death through suppression of mtor signaling pathway in prostate cancer cells. Carcinogenesis 31: 1424-1433.
- Tauro M, Laghezza A, Loiodice F, Piemontese L, Caradonna A, et al. (2016) Catechol-based matrix metalloproteinase inhibitors with additional antioxidative activity. Journal of Enzyme Inhibition and Medicinal Chemistry 31: 25-37.
- Spicer TP, Jiang J, Taylor AB, Choi JY, Hart PJ, et al. (2014) Characterization of selective exosite-binding inhibitors of matrix metalloproteinase 13 that prevent articular cartilage degradation in vitro. Journal of Medicinal Chemistry 57: 9598.
- Zhou S, Klaunig JE (2016) Interplay between mmp-8 and tgf-β1 and its role in regulation of epithelial to mesenchymal transition in hepatocellular carcinoma. Translational Cancer Research 5: S1135-S1138.
- Miron A, Aprotosoaie AC, Trifan A, Xiao J (2017) Flavonoids as modulators of metabolic enzymes and drug transporters. Annals of the New York Academy of Sciences.
- Liu S, Zheng H, Sun R, Jiang H, Chen J, et al. (2017) Disposition of flavonoids for personal intake. Current Pharmacology Reports 3: 196-212.


