Keywords
Harmane, Agmatine, β-carboline, CDS, Imidazoleacetic acid ribotide, Imidazoline binding sites
The Heterogeneity of Imidazoline Binding Sites
The imidazoline binding sites (I-BS) represent a heterogenous family of receptors/sites that stemmed from studies investigating the hypotensive effect of the imidazoline compound clonidine [1]. Structure-affinity relationship studies complemented by functional analyses characterised three types of I-BS, denoted I1-BS, I2-BS (I2A and I2B) and I3-BS [2]. Extensive research has established the involvement of central I1-BS in cardiovascular function [3]. Other reported functions include alleviating symptoms associated with metabolic syndrome X [4] and Huntington’s Disease [5], promoting natriuresis [6], regulating intraocular pressure [7], and modulating mRNA expression for phenylethanolamine N-methyl transferase (PNMT) [8]. Furthermore, the expression of these sites was shown to be altered in conditions such as depression [9] and dysphoric premenstrual syndrome [10] suggesting their involvement in psychiatric conditions and endocrine function respectively. The current literature advocates the role of I2- BS in a number of mood conditions, including depression [11] and anxiety [12]. Furthermore, a strong body of evidence has demonstrated that I2-BS ligands modulate neuropathic pain and analgesia [13-15]. These sites have also been implicated to play a role in attenuating symptoms associated with drug addiction [16], thermoregulation [17], acting as biomarkers for various neurodegenerative diseases and glial tumours [18] and appetite regulation [19]. Lastly, upon activation I3-BS have been shown to exhibit insulinotropic activity as well as inhibit glucagon secretion from pancreatic cells implicating their potential importance in the management of type II diabetes [20].
The anatomical structures of I-BS remain inconclusive, as research has yet to verify the molecular nature of their binding proteins. A protein termed imidazoline receptor antisera selected (IRAS) was reported to represent the I1-BS protein [21,22]. Later, a murine homologue of IRAS Nischarin was cloned [23] illustrating comparable results in biochemical and functional studies that are representative of I1-BS [24-26]. The mitochondrial enzyme monoamine oxidase (MAO) is the most notably sought protein candidate for I2-BS [27,28]. However, various reports have illustrated that only a subset of I2-BS reside on MAO, whilst a fraction of these sites were also found to be associated with brain creatine kinase [29]. Lastly, I3-BS have been proposed to associate with the ion-conducting component Kir 6.2 coupled to KATP channels [30,31].
Proposed endogenous ligands at imidazoline binding sites
The existence of novel sites sensitive to imidazoline-related molecules raises the question as to whether an endogenous modulator does exist. Several candidates have been put forth based on their endogenous occurrence in man, affinity for these sites and their biological activity exerted via I-BS. The leading endogenous candidates include agmatine, imidazoleacetic acid ribotide and harmane (Figure 1).
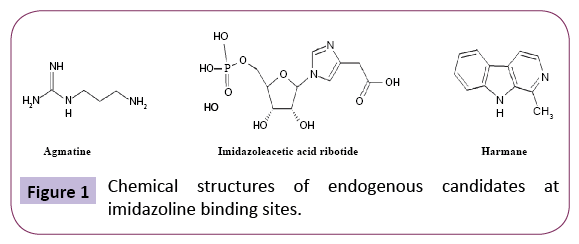
Figure 1: Chemical structures of endogenous candidates at imidazoline binding sites.
The endogenous extract at imidazoline binding sites: clonidine displacing substance
In 1984, Atlas and Burstein isolated and partially purified an endogenous extract from rat and bovine brain homogenates. This extract was termed Clonidine Displacing Substance (CDS) due to its ability to displace specific [3H] clonidine binding in rat brain membrane [32]. CDS was reported to be most abundant in bovine lung [33]. Initially, CDS was proposed to be an endogenous modulator for α2-adrenoceptor (α2-AR) due to its affinity for these sites and its ability to mimic the actions of clonidine: brain CDS extract was shown to promote platelet aggregation and inhibit contractile response in rat vas deferens [34,35]. Conversely, the contractile response induced by serum CDS extracts in aortic tissue preparations was not blocked by the selective α2-AR antagonist rauwolscine [36]; illustrating that CDS does not exert its effect via α2-AR. Interestingly, whilst some groups reported that CDS elicits opposing effects to clonidine, others demonstrated the lack of clonidine-like properties of CDS [37,38]. The reported differences in CDS-evoked responses may be due to the variation in extraction techniques used to isolate and purify CDS, as well as the use of various tissue sources that has led to the ambiguity of its definitive role. CDS extracts from different tissue sources may not contain the same composition of active constituents that are present elsewhere which may account for differences in CDS activity. This notion was addressed by Pinthong et al. demonstrating that CDS extract derived from bovine brain and lung differ in activity; brain CDS attenuated the forskolin-stimulated cyclic adenosine monophosphate (cAMP) accumulation response in guinea pig cerebral cortical preparations whilst lung CDS extract caused an elevation in response [39]. The latter effect was proposed to be mediated by histamine present in lung CDS extract [39]. The chemical composition of CDS is still a matter of debate due to the discrepancies in extraction procedures and the sources of CDS extracts. However, what is currently known about the chemical entity of CDS is that it does not resemble that of a catecholamine nor a peptide [40]. Various groups have identified a number of contaminants present in CDS extracts that have survived the extraction process, including biogenic amines and complex β-carbolines [41,42].
Binding analyses have shown that CDS displays affinity at I1- BS in the rostral ventrolateral medulla (RVLM) and adrenal chromaffin cells [43], and at I2-BS in rat brain membranes [44]. In cardiovascular studies, microinjection of CDS into rat RVLM potentiated a decrease in arterial blood pressure [45] similar to clonidine [1]. However, whether the hypotensive effect of clonidine is mediated solely by α2-AR or I1-BS alone is still a matter of controversy. In contrast, Atlas’ group reported an elevation in mean blood pressure following CDS treatment in cats [46] and rabbits [47]. The hypotensive effect elicited by clonidine was antagonised by CDS [46], suggesting that the component(s) in CDS extract interact at the same receptor as clonidine. In 1997, Chan and her colleagues demonstrated that rat brain-derived CDS extract stimulated glucose-dependent insulin secretion in rat and human isolated islets of Langerhans in a similar manner to efaroxan through its proposed interaction at atypical I3-BS [48]. More importantly this was the first study to show that the imidazoline compounds, RX801080 and KU14R (I3-BS antagonists), could block the secretagogue action of CDS [48]. At present, there has been no evidence to report the action of CDS extract on I2-BS mediated effects. The endogenous expression of component(s) found in the CDS extract displaying affinity and functional activity at I-BS implies that a neuromodulator potentially exists for these sites, and various active components of CDS have been identified and were shown to elicit actions attributable to I-BS involvement.
Agmatine - the endogenous ligand at imidazoline binding sites?
Following the identification of an extract that was shown to display affinity and activity at I-BS it was crucial to isolate the principle component(s) associated with its pharmacology at these sites. In 1994, Li et al. extracted the decarboxylated product of L-arginine, namely agmatine, using ion and molecular weight exclusion chromatography followed by HPLC and mass spectrometry [49]. The presence of agmatine in purified extracts of CDS was shown to display affinity at α2-AR and I-BS, albeit weakly at the latter sites (Ki at I1-BS=30 mM, Ki at I2-BS=100 mM) [50]. Agmatine is synthesised enzymatically from arginine by membrane-bound mitochondrial arginine decarboxylase and is metabolised by agmatinase and/ or diamineoxidase [38]. In neuronal preparations, agmatine was shown to exhibit neurotransmitter-like properties including vesicular storage and extrasynaptic release [51,52]. Furthermore, the cellular uptake of agmatine into mammalian cells was shown to be driven by the organic cation transporter 2 [53]. Thus, it is evident that agmatine fulfils a number of criteria to classify it as a neurotransmitter.
The pharmacological actions of agmatine have been thoroughly documented and shown to mimic some but not all of the properties of CDS. For instance, Regunathan et al. showed that agmatine induced the release of catecholamine from bovine chromaffin cells similarly to CDS [43]. In contrast, cardiovascular studies demonstrated that central administration of agmatine into the RVLM did not reproduce the central actions of clonidine on arterial blood pressure [54]. In behavioural studies, agmatine was shown to promote anticompulsive-like effects similarly to I1- BS and I2-BS selective ligands and was blocked by their respective I-BS antagonists [55]. Moreover, intracerebroventricular administration of agmatine in mice and rats produced an elevation in morphine, ethanol and nicotine induced antinociception; which was also observed with agmatine and the selective I2-BS agonists 2-BFI [15,56,57] and more notably CR4056, which is currently underway for Phase II clinical trials [15]. Furthermore, the antinociceptive effect of agmatine was reversed by idazoxan and BU224; illustrating I2-BS antagonism [56]. The exact method by which I2-BS selective compounds potentiate opioid analgesia is yet uncertain. Studies have shown that I2-BS ligands partly exert their effects through modulation of MAO activity [58]. In 1976, Iwamoto et al. demonstrated that acute treatment of mice with pargyline (irreversible non-selective MAO inhibitor) induced morphine antinociception in the tail-flick test; suggesting MAO involvement [59]. Moreover, morphine along with idazoxan (the first prototypical drug used to assess I2-BS pharmacology) was shown to inhibit MAOB activity in rat brain [60]; which further infers the role of MAO in morphine analgesia. However, it is unlikely that agmatine exerts its analgesic effect through MAO inhibition as agmatine did not affect MAOB activity in rat brain homogenates [60]. As with the case of I2-BS agonists, agmatine would appear to exert its analgesic response potentially through association with a subclass of I2-BS that are not present on MAO, of which remains to be explored.
The current literature proposes that I1-BS and I2-BS play a role in major depression [61]. The expression of I1-BS and I2-BS were reported to be altered in platelets and brains of depressed patients [62-65]. In addition, the immunoreactivity of the I1-BS protein candidate IRAS, as well as the proposed 29/30 kD I2- BS protein, were also notably changed in platelets and brains of depressed suicide victims in comparison to healthy subjects [66,67]. In preclinical studies, selective I2-BS compounds such as 2-BFI and BU224 exhibited antidepressant activity by regulating central monoamine levels [68,69] and increasing mobility time in the Porsolt forced swim paradigm [11,70,71]. Behaviour analyses showed that agmatine along with I-BS ligands (2-BFI, moxonidine and clonidine) mediated the antidepressant like effects of selective serotonin reuptake inhibitors (SSRIs). Furthermore, mice pretreated with SSRIs and exposed to the forced swim test showed overall elevated brain agmatine levels [72]. However, pretreatment of mice with an I-BS antagonist did not block the increase in brain agmatine concentration [72]. Nevertheless, the reported findings show that there is an apparent role of agmatine in mood conditions, which may in part be mediated by I-BS. Agmatine has also been shown to decrease plasma glucose levels in streptozotocin-induced diabetic rats and its lowering actions was hindered following pretreatment using the I2-BS selective ligand BU224 [73]. The insulinotropic activity of agmatine was demonstrated to be less efficacious than the I3-BS ligand efaroxan [74]. Agmatine was shown to block KATP channels in isolated pancreatic β- cells [75] similarly to that illustrated by I3-BS ligands [76].
All in all, does agmatine represent the bioactive component of CDS? In view of the current literature it appears that agmatine and CDS are two separate entities. The physiochemical properties of agmatine differ from that of CDS and the distribution of agmatine does not overlap with that of CDS [38]. Nonetheless, the emergent publications have proposed that agmatine does exert some of its effects through I-BS. However, its low affinity along with its weak activity at I-BS, in comparison to selective I-BS compounds, raises doubt on its fidelity for these sites. Agmatine has been reported to interact with other target sites including N-methyl-D-aspartate (NMDA) receptor [77], 5-HT3 receptor channel [78], nicotinic receptor [79], which in turn may explain its eclectic biological effects.
Imidazoleacetic acid ribotide - The endogenous ligand at imidazoline binding sites?
Another proposed candidate is the phosphoribosylated derivative of imidazoleacetic acid (IAA). IAA is naturally occurring in brain, cerebrospinal fluid and plasma and has been suggested to play a role in brain function [80]. Histamine and histidine were reported to act as precursors for the enzymatic formation of IAA, with the latter being the most predominant pathway [80] (Figure 2). Early research by Ernsberger’s group (1992) demonstrated that IAA displaced [3H] p-aminoclonidine binding from I1-BS at micromolar concentrations [81]. IAA administration into the lateral ventricle of the brain in cats elicited a hypotensive response [82]; corresponding to an I1-BS mediated function [3]. The conjugate metabolite of IAA, namely IAA - ribotide (IAA-RP), was also reported to be present endogenously in rat tissue extracts and in human cerebrospinal fluid [83]. Immunostaining techniques detected the presence of IAA-RP in rat RVLM [83]; a region rich in I1-BS [84]. In synaptosomal preparations, IAA-RP release was elicited by depolarizing concentrations of K+ and its release was noted to be Ca2+-dependent, a feature shared with numerous neurotransmitters [83]. Functional studies demonstrated that IAA-RP promoted arachidonic acid release from PC12 cells [83], a component associated in the signalling pathway for I1- BS [85]. In rat brain, IAA-RP was reported to inhibit excitatory synaptic transmission [86,87]. Treatment with I1-BS antagonist was shown to reverse the synaptic depression induced by IAARP, whilst rauwolscine (α2-AR antagonist) was ineffective [86]. These studies infer that IAA-RP regulates brain synaptic currents through I1-BS involvement but not α2-AR. Further evidence to support the interaction of IAA-RP with I-BS showed that IAA-RP stimulated insulin secretion, which corresponds to I3-BS function, and interestingly IAA-RP elicited a hypertensive response post microinjection in rat RVLM [83]. If IAA-RP were to represent the endogenous ligand for I-BS it would be assumed that it would act as an agonist at these sites. However, the activity of IAA-RP in cardiovascular studies does not correlate to the profile of I1-BS agonists. In addition, the low binding affinity of IAA-RP at I1-BS (Ki=13μM) in bovine adrenal medulla membranes [83] suggests that this compound is a weak endogenous candidate for I1-BS. Moreover, there has been no report on the action of IAA-RP on I2- BS - mediated functions and therefore the speculation of IAA-RP as an endogenous I-BS ligand remains inconclusive.
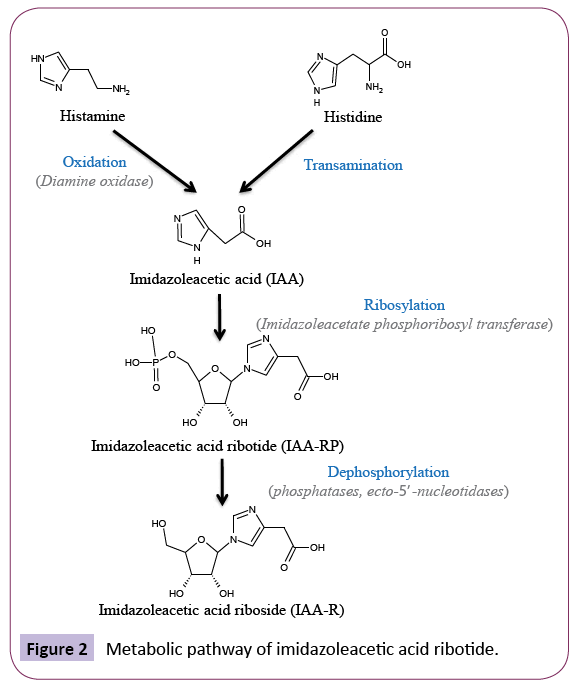
Figure 2: Metabolic pathway of imidazoleacetic acid ribotide.
Harmane - the endogenous ligand at imidazoline binding sites?
One of the latest additions to the list of proposed endogenous molecules at I-BS is the β-carboline harmane [88]; which was reported to be the active constituent of CDS [41]. Harmane and other β-carbolines are naturally occurring in many plants and mammals [89]. In the mammalian body, β-carbolines are formed in a condensation reaction between indolealkylamines and aldehydes [90]. Recent advances have demonstrated that haem peroxidases catalyse the oxidation of (tetrahydro-β-carbolines) THβC to form harmane and norharmane [91]. Furthermore, these β-carbolines were shown to be metabolised by subtypes of cytochrome P450 enzymes [92] (Figure 3). Recent studies have shown that harmane accumulation in rat brain cortex does not represent an active-driven mechanism [93]. Furthermore, extracellular depolarizing concentrations of K+ failed to induce the release of [3H]harmane from brain cortical tissue; suggesting that harmane does not represent a classical neurotransmitter [93].
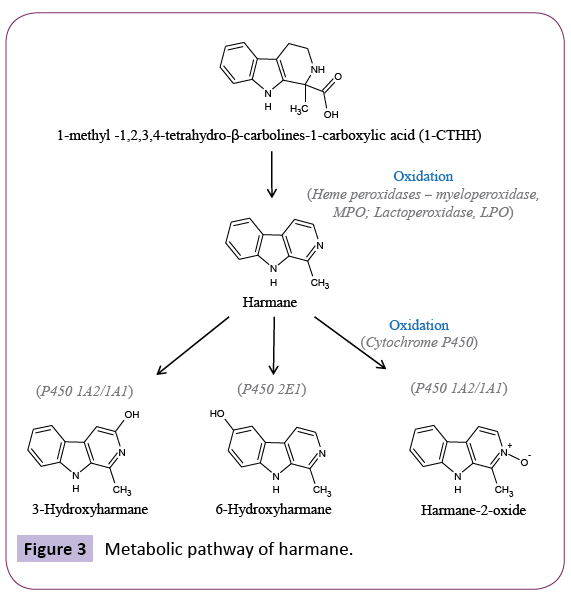
Figure 3: Metabolic pathway of harmane.
Harmane has been shown to interact with different classes of receptors including serotonin, dopamine, benzodiazepine, histamine and nicotine [94-96]. In addition, harmane has also been shown to interact with I-BS with a higher affinity (IC50 at I1-BS=31 nM, Ki at I2-BS=49 nM) [97] compared to agmatine. The natural occurrence of harmane in the arcuate nucleus [98], a region rich in both I2-BS [99] and MAOs [100], could suggest a possible role for harmane as an endogenous ligand at these sites.
Functional studies demonstrated that administration of harmane into rat brain RVLM elicited a hypotensive response which was also observed with I1-BS ligands clonidine, rilmenidine, moxonidine and LNP 509 [101,102]. Furthermore, the central antihypertensive action of harmane and clonidine was reversed by the mixed α2-AR/I1-BS antagonist efaroxan. However, efaroxan was more efficacious against harmane than clonidine [101]. The hypotensive effect elicited by harmane is in agreement with the functional role of I1-BS in the regulation of blood pressure at the level of the brainstem [3]. These findings along with the structure-affinity relationship of harmane binding to I1-BS propose that harmane is a ligand at I1-BS. In the rat central nervous system (CNS), harmane has been shown to modulate monoamine turnover [103] in a comparable manner to selective I2-BS ligands such as 2-BFI and BU224 [68,70]. The regulation of central monoamine levels by harmane and I2-BS compounds was proposed to occur partially through MAO inhibition [104]. Brain map imaging demonstrated that [3H]harmane distribution correlated with I2-BS and MAOA distribution [105,106], further strengthening the mode of action of harmane on these sites. In more recent studies, harmane was reported to elicit the spontaneous efflux of 5-HT from rat brain cortex devoid of MAO involvement [107]. Thus, these studies highlight that in addition to MOA inhibition harmane can partially regulate brain monoamine levels by inducing synaptic release.
A vast amount of research has explored the pharmacology of harmane at I-BS. Canular infusion of harmane into the dorsal hippocampus has been shown to stimulate feeding in rats [108] and this phenomenon was similarly documented with selective I2-BS ligands [109]. Harmane has also been shown to induce hypothermia in rats partially through MAO inhibition [110]. The homeostatic regulation of body temperature was also reported by high affinity ligands at I2-BS namely 2-BFI, BU224 and CR4056 [17]; whilst no effect was observed by agmatine, another candidate endogenous ligand at I-BS [111]. Thus, it is likely that harmane potentiates its hypothermic response via I2-BS association. In pancreatic β-cells, harmane induced insulin release through a proposed action at I3-BS, a site known to regulate insulin secretion [20,112]. Although, ryanodine receptors and L-type calcium channels have also been suggested to play a role in mediating the insulinotropic activity of harmane [112]. The insulin secretagogue activity of harmane is analogous to that of CDS and efaroxan, such that they all antagonised the inhibitory response of diazoxide (KATP agonist) on glucose-mediated insulin release [113]. Furthermore, KU14R (I3-BS antagonist) decreased the stimulatory effect of both harmane and efaroxan implicating that harmane mediates the secretion of insulin via a similar mechanism of action as efaroxan [113].
In behavioural studies, harmane has been shown to alleviate some symptoms associated with drug abuse and tolerance, which is thought to be mediated by I-BS [114]. Similarly, agmatine, BU224 and clonidine had also been reported to attenuate many behavioural characteristics of morphine abstinence syndrome [115]. In drug-discrimination models, harmane along with selective I2-BS compounds were shown to potently and dose-dependently substitute for 2-BFI in the twolever operant chamber model [116]. Moreover, harmane was also reported to substitute BU224 in drug discrimination studies [117]. The ability of harmane, along with I2-BS agonists such as CR4056, to substitute for BU224 (I2-BS antagonist) in discrimination studies is somewhat puzzling and requires further exploration. Previous approaches demonstrated that idazoxan also substituted for 2-BFI, but clonidine did not; illustrating specificity to I2-BS interaction and not I1-BS [118]. Interestingly, idazoxan displays antagonistic activity at I2-BS. Therefore, it would have been expected that the antagonistic profile of idazoxan would attenuate rather than promote the discriminative stimulus effects. It was proposed that idazoxan behaves as a partial agonist at I2-BS in systems that require low efficacy demand, in the case of drug discrimination, and would act as a full antagonist in assays that require higher efficacy demand, such as pain modulation [119]. In more recent studies, Qiu et al. reported that I2-BS ligands, including the proposed I-BS endogenous ligand harmane, substituted for CR4056 (high affinity I2-BS ligand) in the two-lever drug-discrimination model [119]. Although harmane at 10mg/kg only displayed partial substitution, it was more potent than agmatine as it failed to substitute for CR4056 at the same dose [119].
Preclinical studies have shown that harmane exhibits anxiolytic and anti-depressant properties [120], which was also projected by I2-BS ligands [11,70,121]. In restraint-stressed rats, harmane and BU224 treatment elevated Fos expression in distinct brain regions in a comparable manner; emphasizing the overlap of these molecules for the same target site [12]. Recently, Aglawe et al. described the anti-nociceptive activity of harmane to that of I1- BS and I2-BS agonists demonstrating similar functional responses [57]. However, whether the antinociceptive effect of harmane is preferentially elicited through its action on I1-BS alone, I2-BS alone or both needs to be addressed.
In light of the current literature it appears that harmane represents a strong candidate as an endogenous ligand at I-BS due to its high affinity for these sites and its ability to reproduce various effects that have been noted to occur by selective I-BS compounds. However, in order to truly crown harmane as an endogenous molecule its neurotransmitter properties need to be addressed to confirm its credibility to earn the title as a neurotransmitter/neuromodulator.
Conclusion
The emergent information on I-BS has shed light on their functional significance and importance in an array of medical conditions. The current review discusses the pharmacology of agmatine, imidazoleacetic acid ribotide and harmane as putative endogenous compounds at I-BS, reporting their functional overlap to known selective molecules for these sites. However, the lack of a strong protein candidate for these sites has made it difficult to characterize the true identity of the endogenous ligand. Moreover, the discrepancies in the purification procedures and the controversial findings of some of the effects of these ligands need to be resolved in order to unravel the “borne identity” of the molecule for I-BS.
6601
References
- Bousquet P, Feldman J, Schwartz J (1984) Central cardiovascular effects of alpha adrenergic drugs: differences between catecholamines and imidazolines. J Pharmacol Exp Ther 230: 232-236.
- Eglen RM, Hudson AL, Kendall DA, Nutt DJ, Morgan NG, et al. (1998) 'Seeing through a glass darkly': casting light on imidazoline 'I' sites. Trends Pharmacol Sci 19: 381-390.
- Bousquet P, Dontenwill M, Greney H, Feldman J (2000) Imidazoline receptors in cardiovascular and metabolic diseases. J Cardiovasc Pharmacol 35: S21-25.
- Velliquette RA, Kossover R, Previs SF, Ernsberger P (2006) Lipid-lowering actions of imidazoline antihypertensive agents in metabolic syndrome X. Naunyn Schmiedebergs Arch Pharmacol 372: 300-312.
- Gupta S, Sharma B (2014) Pharmacological benefit of I(1)-imidazoline receptors activation and nuclear factor kappa-B (NF-κB) modulation in experimental Huntington's disease. Brain Res Bull 102: 57-68.
- Fauvel JP, Najem R, Ryon B, Ducher M, Laville M (1999) Effects of rilmenidine on stress-induced peak blood pressure and renal function. J Cardiovasc Pharmacol 34: 41-45.
- Campbell WR, Potter DE (1994) The central effects of moxonidine on intraocular pressure and its antagonism by L-659, 066 and L-657, 743 in the rabbit. Prog Neuropsychopharmacol Biol Psychiatry 18: 1051-1061.
- Evinger MJ, Ernsberger P, Regunathan S, Reis DJ (1995) Regulation of phenylethanolamine N-methyltransferase gene expression by imidazoline receptors in adrenal chromaffin cells. J Neurochem 65: 988-997.
- Piletz JE, Halaris AE, Chikkala D, Qu Y (1996) Platelet I1-imidazoline binding sites are decreased by two dissimilar antidepressant agents in depressed patients. J Psychiatr Res 30: 169-184.
- Halbreich U, Piletz JE, Carson S, Halaris A, Rojansky N (1993) Increased imidazoline and alpha 2 adrenergic binding in platelets of women with dysphoric premenstrual syndromes. Biol Psychiatry 34: 676-686.
- Nutt DJ, French N, Handley S, Hudson A, Husbands S, et al. (1995) Functional studies of specific imidazoline-2 receptor ligands. Ann N Y Acad Sci 763: 125-139.
- Smith KL, Roche M, Jessop DS, Finn DP (2009) The effects of synthetic and endogenous imidazoline binding site ligands on neuronal activity in discrete brain regions of naive and restraint-stressed rats. Eur Neuropsychopharmacol 19: 371-380.
- Li JX, Zhang Y (2011) Imidazoline I2 receptors: target for new analgesics? Eur J Pharmacol 658: 49-56.
- Li JX, Thorn DA, Qiu Y, Peng BW, Zhang Y (2014) Antihyperalgesic effects of imidazoline I(2) receptor ligands in rat models of inflammatory and neuropathic pain. Br J Pharmacol 171: 1580-1590.
- Lanza M, Ferrari F, Menghetti I, Tremolada D, Caselli G (2014) Modulation of imidazoline I2 binding sites by CR4056 relieves postoperative hyperalgesia in male and female rats. Br J Pharmacol 171: 3693-3701.
- Miralles A, Esteban S, Sastre-Coll A, Moranta D, Asensio VJ, et al. (2005) High-affinity binding of beta-carbolines to imidazoline I2B receptors and MAO-A in rat tissues: norharman blocks the effect of morphine withdrawal on DOPA/noradrenaline synthesis in the brain. Eur J Pharmacol 518: 234-242.
- Thorn DA, An XF, Zhang Y, Pigini M, Li JX (2012) Characterization of the hypothermic effects of imidazoline Iâ‚‚ receptor agonists in rats. Br J Pharmacol 166: 1936-1945.
- García-Sevilla JA, Escribá PV, Guimón J (1999) Imidazoline receptors and human brain disorders. Ann N Y Acad Sci 881: 392-409.
- Brown CM, MacKinnon AC, Redfern WS, Williams A, Linton C, et al. (1995) RS-45041-190: a selective, high-affinity ligand for I2 imidazoline receptors. Br J Pharmacol 116: 1737-1744.
- Chan SL, Dunne MJ, Stillings MR, Morgan NG (1991) The alpha 2-adrenoceptor antagonist efaroxan modulates K+ATP channels in insulin-secreting cells. Eur J Pharmacol 204: 41-48.
- Piletz JE, Ivanov TR, Sharp JD, Ernsberger P, Chang CH, et al. (2000) Imidazoline receptor antisera-selected (IRAS) cDNA: cloning and characterization. DNA Cell Biol 19: 319-329.
- Ivanov TR, Jones JC, Dontenwill M, Bousquet P, Piletz JE (1998) Characterization of a partial cDNA clone detected by imidazoline receptor-selective antisera. J Auton Nerv Syst 72: 98-110.
- Alahari SK, Lee JW, Juliano RL (2000) Nischarin, a novel protein that interacts with the integrin alpha5 subunit and inhibits cell migration. J Cell Biol 151: 1141-1154.
- Sun Z, Chang CH, Ernsberger P (2007) Identification of IRAS/Nischarin as an I1-imidazoline receptor in PC12 rat pheochromocytoma cells. J Neurochem 101: 99-108.
- Zhang J, Abdel-Rahman AA (2006) Nischarin as a functional imidazoline (I1) receptor. FEBS Lett 580: 3070-3074.
- Zhang J, Abdel-Rahman AA (2008) Inhibition of nischarin expression attenuates rilmenidine-evoked hypotension and phosphorylated extracellular signal-regulated kinase 1/2 production in the rostral ventrolateral medulla of rats. J Pharmacol Exp Ther 324: 72-78.
- Tesson F, Limon-Boulez I, Urban P, Puype M, Vandekerckhove J, et al. (1995) Localization of I2-imidazoline binding sites on monoamine oxidases. J Biol Chem 270: 9856-9861.
- Parini A, Moudanos CG, Pizzinat N, Lanier SM (1996) The elusive family of imidazoline binding sites. Trends Pharmacol Sci 17: 13-16.
- Kimura A, Tyacke RJ, Minchin MC, Nutt DJ, Hudson AL (2003) Identification of an I(2) binding protein from rabbit brain. Ann N Y Acad Sci 1009: 364-366.
- Proks P, Ashcroft FM (1997) Phentolamine block of KATP channels is mediated by Kir6.2. Proc Natl Acad Sci U S A 94: 11716-11720.
- Morgan NG, Chan SL, Mourtada M, Monks LK, Ramsden CA (1999) Imidazolines and pancreatic hormone secretion. Ann N Y Acad Sci 881: 217-228.
- Atlas D, Burstein Y (1984) Isolation and partial purification of a clonidine-displacing endogenous brain substance. Eur J Biochem 144: 287-293.
- Singh G, Hussain JF, MacKinnon A, Brown CM, Kendall DA, et al. (1995) Evidence for the presence of a non-catecholamine, clonidine-displacing substance in crude, methanolic extracts of bovine brain and lung. Naunyn Schmiedebergs Arch Pharmacol 351: 17-26.
- Atlas D, Diamant S, Fales HM, Pannell L (1987) The brain's own clonidine: purification and characterization of endogenous clonidine displacing substance from brain. J Cardiovasc Pharmacol 10 Suppl 12: S122-127.
- Diamant S, Atlas D (1986) An endogenous brain substance, CDS (clonidine-displacing-substance), inhibits the twitch response of rat vas deferens. Biochem Biophys Res Commun 134: 184-190.
- Synetos D, Manolopoulos VG, Atlas D, Pipili-Synetos E (1991) Human plasma-derived material with clonidine displacing substance (CDS)-like properties contracts the isolated rat aorta. J Auton Pharmacol 11: 343-351.
- Pinthong D, Hussain JF, Kendall DA, Wilson VG (1995) Comparison of the interaction of agmatine and crude methanolic extracts of bovine lung and brain with alpha 2-adrenoceptor binding sites. Br J Pharmacol 115: 689-695.
- Raasch W, Schäfer U, Chun J, Dominiak P (2001) Biological significance of agmatine, an endogenous ligand at imidazoline binding sites. Br J Pharmacol 133: 755-780.
- Pinthong D, Kendall DA, Wilson VG (2003) Complex interaction of alpha(2)-adrenoceptor binding sites with bovine brain and lung extracts containing clonidine-displacing substance. Ann N Y Acad Sci 1009: 216-221.
- Atlas D (1994) Identifying clonidine-displacing substance. Science 266: 462-464.
- Parker CA, Anderson NJ, Robinson ES, Price R, Tyacke RJ, et al. (2004) Harmane and harmalan are bioactive components of classical clonidine-displacing substance. Biochemistry 43: 16385-16392.
- Grigg M, Musgrave IF, Barrow CJ (1998) Isolation and partial structure determination of a clonidine-displacing substance from bovine lung and brain. J Auton Nerv Syst 72: 86-93.
- Regunathan S, Meeley MP, Reis DJ (1991) Clonidine-displacing substance from bovine brain binds to imidazoline receptors and releases catecholamines in adrenal chromaffin cells. Mol Pharmacol 40: 884-888.
- Parker CA, Hudson AL, Nutt DJ, Dillon MP, Eglen RM, et al. (1999) Extraction of active clonidine-displacing substance from bovine lung and comparison with clonidine-displacing substance extracted from other tissues. Eur J Pharmacol 378: 213-221.
- Meeley MP, Ernsberger PR, Granata AR, Reis DJ (1986) An endogenous clonidine-displacing substance from bovine brain: receptor binding and hypotensive actions in the ventrolateral medulla. Life Sci 38: 1119-1126.
- Bousquet P, Feldman J, Atlas D (1987) Central cardiovascular effects of a noncatecholamine endogenous ligand for clonidine receptors. J Cardiovasc Pharmacol 10 Suppl 12: S167-171.
- Bousquet P, Feldman J, Atlas D (1986) An endogenous, non-catecholamine clonidine antagonist increases mean arterial blood pressure. Eur J Pharmacol 124: 167-170.
- Chan SL, Atlas D, James RF, Morgan NG (1997) The effect of the putative endogenous imidazoline receptor ligand, clonidine-displacing substance, on insulin secretion from rat and human islets of Langerhans. Br J Pharmacol 120: 926-932.
- Li G, Regunathan S, Barrow CJ, Eshraghi J, Cooper R, et al. (1994) Agmatine: an endogenous clonidine-displacing substance in the brain. Science 263: 966-969.
- Piletz JE, Chikkala DN, Ernsberger P (1995) Comparison of the properties of agmatine and endogenous clonidine-displacing substance at imidazoline and alpha-2 adrenergic receptors. J Pharmacol Exp Ther 272: 581-587.
- Sastre M, Regunathan S, Reis DJ (1997) Uptake of agmatine into rat brain synaptosomes: possible role of cation channels. J Neurochem 69: 2421-2426.
- Goracke-Postle CJ, Nguyen HO, Stone LS, Fairbanks CA (2006) Release of tritiated agmatine from spinal synaptosomes. Neuroreport 17: 13-17.
- Higashi K, Imamura M, Fudo S, Uemura T, Saiki R, et al. (2014) Identification of functional amino acid residues involved in polyamine and agmatine transport by human organic cation transporter 2. PLoS One 9: e102234.
- Sun MK, Regunathan S, Reis DJ (1995) Cardiovascular responses to agmatine, a clonidine-displacing substance, in anesthetized rat. Clin Exp Hypertens 17: 115-128.
- Dixit MP, Thakre PP, Pannase AS, Aglawe MM, Taksande BG, et al. (2014) Imidazoline binding sites mediates anticompulsive-like effect of agmatine in marble-burying behavior in mice. Eur J Pharmacol 732: 26-31.
- Sánchez-Blázquez P, Boronat MA, Olmos G, García-Sevilla JA, Garzón J (2000) Activation of I(2)-imidazoline receptors enhances supraspinal morphine analgesia in mice: a model to detect agonist and antagonist activities at these receptors. Br J Pharmacol 130: 146-152.
- Aglawe MM, Taksande BG, Kuldhariya SS, Chopde CT, Umekar MJ, et al. (2014) Participation of central imidazoline binding sites in antinociceptive effect of ethanol and nicotine in rats. Fundam Clin Pharmacol 28: 284-293.
- Raasch W, Muhle H, Dominiak P (1999) Modulation of MAO activity by imidazoline and guanidine derivatives. Ann N Y Acad Sci 881: 313-331.
- Iwamoto ET, Ho IK, Way EL (1976) Effect of pargyline on morphine tolerance and physical dependence development in mice. Eur J Pharmacol 38: 261-268.
- Su RB, Li J, Li X, Qin BY (2001) Down-regulation of MAO-B activity and imidazoline receptors in rat brain following chronic treatment of morphine. Acta Pharmacol Sin 22: 639-644.
- Halaris A, Piletz JE (2001) Imidazoline receptors: possible involvement in the pathophysiology and treatment of depression. Hum Psychopharmacol 16: 65-69.
- Piletz JE, Halaris A, Saran A, Marler M (1990) Elevated 3H-para-aminoclonidine binding to platelet purified plasma membranes from depressed patients. Neuropsychopharmacology 3: 201-210.
- Piletz JE, Halaris A, Saran A, Marler MR (1991) Desipramine lowers tritiated para-aminoclonidine binding in platelets of depressed patients. Arch Gen Psychiatry 48: 813-820.
- Alemany R, Olmos G, García-Sevilla JA (1995) The effects of phenelzine and other monoamine oxidase inhibitor antidepressants on brain and liver I2 imidazoline-preferring receptors. Br J Pharmacol 114: 837-845.
- Piletz JE, Zhu H, Ordway G, Stockmeier C, Dilly G, et al. (2000) Imidazoline receptor proteins are decreased in the hippocampus of individuals with major depression. Biol Psychiatry 48: 910-919.
- García-Sevilla JA, Escribá PV, Sastre M, Walzer C, Busquets X, et al. (1996) Immunodetection and quantitation of imidazoline receptor proteins in platelets of patients with major depression and in brains of suicide victims. Arch Gen Psychiatry 53: 803-810.
- Ma JK, Zhu HE, Piletz JE (2003) Effect of postmortem delay on imidazoline receptor-binding proteins in human and mouse brain. Ann N Y Acad Sci 1009: 341-346.
- Lalies MD, Nutt DJ (1995) The effect of a selective I2-site ligand, 2-(-2-benzofuranyl)-2-imidazoline, on in vivo noradrenaline release in rat brain. Br J Pharmacol 114: 413.
- Ugedo L, Pineda J, Ruiz-Ortega JA, Martín-Ruiz R (1998) Stimulation of locus coeruleus neurons by non-I1/I2-type imidazoline receptors: an in vivo and in vitro electrophysiological study. Br J Pharmacol 125: 1685-1694.
- Finn DP, Martí O, Harbuz MS, Vallès A, Belda X, et al. (2003) Behavioral, neuroendocrine and neurochemical effects of the imidazoline I2 receptor selective ligand BU224 in naive rats and rats exposed to the stress of the forced swim test. Psychopharmacology (Berl) 167:195-202.
- Hudson AL, Tyacke RJ, Lalies MD, Davies N, Finn DP, et al. (2003) Novel ligands for the investigation of imidazoline receptors and their binding proteins. Ann N Y Acad Sci 1009: 302-308.
- Taksande BG, Kotagale NR, Tripathi SJ, Ugale RR, Chopde CT (2009) Antidepressant like effect of selective serotonin reuptake inhibitors involve modulation of imidazoline receptors by agmatine. Neuropharmacology 57: 415-424.
- Jou SB, Liu IM, Cheng JT (2004) Activation of imidazoline receptor by agmatine to lower plasma glucose in streptozotocin-induced diabetic rats. Neurosci Lett 358: 111-114.
- Berdeu D, Puech R, Loubatières-Mariani MM, Bertrand G (1996) Agmatine is not a good candidate as endogenous ligand for imidazoline sites of pancreatic B cells and vascular bed. Eur J Pharmacol 308: 301-304.
- Shepherd RM, Hashmi MN, Kane C, Squires PE, Dunne MJ (1996) Elevation of cytosolic calcium by imidazolines in mouse islets of Langerhans: implications for stimulus-response coupling of insulin release. Br J Pharmacol 119: 911-916.
- Dunne MJ, Harding EA, Jaggar JH, Squires PE, Liang R, et al. (1995) Potassium channels, imidazolines, and insulin-secreting cells. Ann N Y Acad Sci 763: 243-261.
- Gibson DA, Harris BR, Rogers DT, Littleton JM (2002) Radioligand binding studies reveal agmatine is a more selective antagonist for a polyamine-site on the NMDA receptor than arcaine or ifenprodil. Brain Res 952: 71-77.
- Molderings GJ, Schmidt K, Bönisch H, Göthert M (1996) Inhibition of 5-HT3 receptor function by imidazolines in mouse neuroblastoma cells: potential involvement of sigma 2 binding sites. Naunyn Schmiedebergs Arch Pharmacol 354: 245-252.
- Quik M (1985) Inhibition of nicotinic receptor mediated ion fluxes in rat sympathetic ganglia by BGT II-S1 a potent phospholipase. Brain Res 325: 79-88.
- Tunnicliff G (1998) Pharmacology and function of imidazole 4-acetic acid in brain. Gen Pharmacol 31: 503-509.
- Ernsberger PL, Westbrooks KL, Christen MO, Schafer SG (1992) A second generation of centrally acting antihypertensive agents act on putative I1-imidazoline receptors. J Cardiovasc Pharmacol 20: 1-10.
- Walland A (1975) cAMP as a second messenger in central blood pressure control. Naunyn Schmiedebergs Arch Pharmacol 290: 419-423.
- Prell GD, Martinelli GP, Holstein GR, Matulić-Adamić J, Watanabe KA, et al. (2004) Imidazoleacetic acid-ribotide: an endogenous ligand that stimulates imidazol(in)e receptors. Proc Natl Acad Sci U S A 101: 13677-13682.
- Ernsberger P, Graves ME, Graff LM, Zakieh N, Nguyen P, et al. (1995) I1-imidazoline receptors. Definition, characterization, distribution, and transmembrane signaling. Ann N Y Acad Sci 763: 22-42.
- Ernsberger P (1998) Arachidonic acid release from PC12 pheochromocytoma cells is regulated by I1-imidazoline receptors. J Auton Nerv Syst 72: 147-154.
- Bozdagi O, Wang XB, Martinelli GP, Prell G, Friedrich VL Jr, et al. (2011) Imidazoleacetic acid-ribotide induces depression of synaptic responses in hippocampus through activation of imidazoline receptors. J Neurophysiol 105: 1266-1275.
- Martir JF, Bozdagi O, Martinelli GP, Friedrich VL, Holstein GR (2012) Imidazoleacetic acid-ribotide in the rodent striatum: a putative neurochemical link between motor and autonomic deficits in Parkinson's disease. Acta Biol Hung 63 Suppl 1: 5-18.
- Hudson AL, Price R, Tyacke RJ, Lalies MD, Parker CA, et al. (1999) Harmane, norharmane and tetrahydro ß-carboline have high affinity for rat imidazoline binding sites. Br J Pharmacol 126: 2.
- Airaksinen MM, Kari I (1981) Beta-carbolines, psychoactive compounds in the mammalian body. Part I: Occurrence, origin and metabolism. Med Biol 59: 21-34.
- McIsaac WM, Taylor D, Walker KE, Ho BT (1972) 6-Methoxy-,,,4-tetrahydro- -carboline--a serotonin elevator. J Neurochem 19: 1203-1206.
- Herraiz T, Galisteo J (2014) Naturally-occurring tetrahydro-β-carboline alkaloids derived from tryptophan are oxidized to bioactive β-carboline alkaloids by heme peroxidases. Biochem Biophys Res Commun 451: 42-47.
- Herraiz T, Guillén H, Arán VJ (2008) Oxidative metabolism of the bioactive and naturally occurring beta-carboline alkaloids, norharman and harman, by human cytochrome P450 enzymes. Chem Res Toxicol 21: 2172-2180.
- Abu Ghazaleh H, Lalies MD, Nutt DJ, Hudson AL (2015) Harmane: an atypical neurotransmitter? Neurosci Lett 590: 1-5.
- Glennon RA, Dukat M, Grella B, Hong S, Costantino L, et al. (2000) Binding of beta-carbolines and related agents at serotonin (5-HT(2) and 5-HT(1A)), dopamine (D(2)) and benzodiazepine receptors. Drug Alcohol Depend 60: 121-132.
- Nasehi M, Mashaghi E, Khakpai F, Zarrindast MR (2013) Suggesting a possible role of CA1 histaminergic system in harmane-induced amnesia. Neurosci Lett 556: 5-9.
- Arib O, Rat P, Molimard R, Chait A, Faure P, et al. (2010) Electrophysiological characterization of harmane-induced activation of mesolimbic dopamine neurons. Eur J Pharmacol 629: 47-52.
- Husbands SM, Glennon RA, Gorgerat S, Gough R, Tyacke R, et al. (2001) beta-carboline binding to imidazoline receptors. Drug Alcohol Depend 64: 203-208.
- Shoemaker DW, Cummins JT, Bidder TG, Boettger HG, Evans M (1980) Identification of harman in the rat arcuate nucleus. Naunyn Schmiedebergs Arch Pharmacol 310: 227-230.
- Lione LA, Nutt DJ, Hudson AL (1998) Characterisation and localisation of [3H]2-(2-benzofuranyl)-2-imidazoline binding in rat brain: a selective ligand for imidazoline I2 receptors. Eur J Pharmacol 353: 123-135.
- Saura J, Kettler R, Da Prada M, Richards JG (1992) Quantitative enzyme radioautography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: localization and abundance of MAO-A and MAO-B in rat CNS, peripheral organs, and human brain. J Neurosci 12: 1977-1999.
- Musgrave IF, Badoer E (2000) Harmane produces hypotension following microinjection into the RVLM: possible role of I(1)-imidazoline receptors. Br J Pharmacol 129: 1057-1059.
- Estato V, Araújo CV, Bousquet P, Tibiriçá E (2004) Effects of centrally acting antihypertensive drugs on the microcirculation of spontaneously hypertensive rats. Braz J Med Biol Res 37: 1541-1549.
- Smith KL, Ford GK, Jessop DS, Finn DP (2013) Behavioural, neurochemical and neuroendocrine effects of the endogenous β-carboline harmane in fear-conditioned rats. J Psychopharmacol 27: 162-170.
- Glover V, Liebowitz J, Armando I, Sandler M (1982) beta-Carbolines as selective monoamine oxidase inhibitors: in vivo implications. J Neural Transm 54: 209-218.
- Anderson NJ, Seif I, Nutt DJ, Hudson AL, Robinson ES (2006) Autoradiographical distribution of imidazoline binding sites in monoamine oxidase A deficient mice. J Neurochem 96: 1551-1559.
- Anderson NJ, Tyacke RJ, Husbands SM, Nutt DJ, Hudson AL, et al. (2006) In vitro and ex vivo distribution of [3H]harmane, an endogenous beta-carboline, in rat brain. Neuropharmacology 50: 269-276.
- Abu Ghazaleh H, Lalies MD, Nutt DJ, Hudson AL (2015) The modulatory action of harmane on serotonergic neurotransmission in rat brain. Brain Res 1597: 57-64.
- Adell A, Myers RD (1994) Increased alcohol intake in low alcohol drinking rats after chronic infusion of the beta-carboline harman into the hippocampus. Pharmacol Biochem Behav 49: 949-953.
- Polidori C, Gentili F, Pigini M, Quaglia W, Panocka I, et al. (2000) Hyperphagic effect of novel compounds with high affinity for imidazoline I(2) binding sites. Eur J Pharmacol 392: 41-49.
- Adell A, Biggs TA, Myers RD (1996) Action of harman (1-methyl-beta-carboline) on the brain: body temperature and in vivo efflux of 5-HT from hippocampus of the rat. Neuropharmacology 35: 1101-1107.
- Bhalla S, Andurkar SV, Gulati A (2013) Involvement of α₂-adrenoceptors, imidazoline, and endothelin-A receptors in the effect of agmatine on morphine and oxycodone-induced hypothermia in mice. Fundam Clin Pharmacol 27: 498-509.
- Squires PE, Hills CE, Rogers GJ, Garland P, Farley SR, et al. (2004) The putative imidazoline receptor agonist, harmane, promotes intracellular calcium mobilisation in pancreatic beta-cells. Eur J Pharmacol 501: 31-39.
- Cooper EJ, Hudson AL, Parker CA, Morgan NG (2003) Effects of the beta-carbolines, harmane and pinoline, on insulin secretion from isolated human islets of Langerhans. Eur J Pharmacol 482: 189-196.
- Aricioglu-Kartal F, Kayir H, Tayfun Uzbay I (2003) Effects of harman and harmine on naloxone-precipitated withdrawal syndrome in morphine-dependent rats. Life Sci 73: 2363-2371.
- Hudson AL, Gough R, Tyacke R, Lione L, Lalies M, et al. (1999) Novel selective compounds for the investigation of imidazoline receptors. Ann N Y Acad Sci 881: 81-91.
- MacInnes N, Handley SL (2002) Characterization of the discriminable stimulus produced by 2-BFI: effects of imidazoline I(2)-site ligands, MAOIs, beta-carbolines, agmatine and ibogaine. Br J Pharmacol 135: 1227-1234.
- Qiu Y, Zhang Y, Li JX (2015) Discriminative stimulus effects of the imidazoline I2 receptor ligands BU224 and phenyzoline in rats. Eur J Pharmacol 749: 133-141.
- Jordan S, Jackson HC, Nutt DJ, Handley SL (1996) Discriminative stimulus produced by the imidazoline I2 site ligand, 2 -BFI. J Psychopharmacol 10: 273-278.
- Qiu Y, He XH, Zhang Y, Li JX (2014) Discriminative stimulus effects of the novel imidazoline Iâ‚‚ receptor ligand CR4056 in rats. Sci Rep 4: 6605.
- Aricioglu F, Altunbas H (2003) Harmane induces anxiolysis and antidepressant-like effects in rats. Ann N Y Acad Sci 1009: 196-201.
- Hudson AL, Nutt DJ, Husbands SM (2001) Imidazoline receptors and their role in depression. Pharm News 2001.

