Keywords
Cannabinoids, ACEA, AM251, dopaminergic neurons, cell death
Introduction
The endocannabinoid system is present in all mammals, widespread for the whole body with different functions. As known, CB1 and CB2 receptors, endogenous ligands (anandamide and 2-aracdonoyl-glycerol, 2-AG; the two most studied), and their synthetic and degradation enzymes compose this system [1,2].
In the nervous system, the cannabinoids signaling has been implicated in several processes both in healthy and pathological brain. A retrograde signaling of endocannabinoid neurotransmission is suggested, especially by the predominant presynaptic localization of cannabinoid receptors in gabaergic and glutamatergic terminals, but also in other “classical” neurotransmitter neurons [3-8].
In addition to its participation in neuroplasticity mechanisms, the endocannabinoid system has been implicated in neurodegenerative processes and different types of experimental approaches have also suggested its neuroprotective properties [4-7]. Accordingly, many efforts have been made over the past 20 years in search to learn how the endocannabinoid system work in our brain and we are still seeking the better way to manipulate this system as therapy to prevent or even rehabilitate some diseases such as Parkinson's disease, Alzheimer's disease, and Multiple Sclerosis [8].
Increased and decreased levels of endocannabinoids and changes in the expression of CB1 receptors in brain damage models in rats and mice have been demonstrated [9-16]. Besides, in human brain of patients with neurodegenerative diseases, such as Huntington’s and Parkinson’s disease [17,18] have also been shown altered levels of endocannabinoid, despite that, cannabinoids have provided symptomatic relief in experimental models of chronic neurodegenerative diseases [19].
In this context, the high density of CB1 receptors and endocannabinoids in the basal ganglia [20-22] has been explored in terms of prospects for therapies to Parkinson’s disease (PD), a neurodegenerative disorder characterized by the loss of dopaminergic basal ganglia neurons. For instance, Delta 9-tetrahydrocannabinol (delta 9-THC) was able to reverse the decreased dopaminergic transmission in the basal ganglia of a mice model of PD [23]. Our group have previously shown, for example, a complex and time-dependent profile of CB1 expression in the basal ganglia nuclei of rat experimental model of PD, produced by intrastriatal 6-hydroxydopamine injections [24], suggesting a participation of cannabinoid system in compensatory mechanisms of neuronal plasticity. However, no studies have been done in in vitro dopaminergic cells for the comprehension of the mechanisms behind synthetic cannabinoid actions.
Notwithstanding, several studies suggest a neuroprotective role of cannabinoid system in neurodegenerative conditions, the data are still conflicting and the mechanisms are largely unknown. The aim of this study was to evaluate whether the cannabinoid compounds (ACEA and AM251, CB1 receptor ligands) would be able to protect the neuroblastoma cell line (Neuro 2a), differentiated into dopaminergic cells, against three different types of damages: 6-hydroxydopamine, lipopolysaccharide, and hydrogen peroxide for 24 hours. These agents were chosen to reproduce the neurotoxic, inflammatory and oxidative parameters, respectively, related to neurodegenerative conditions. We also evaluated the effects of cannabinoid treatment upon the reactive oxygen species production (ROS).
Methods
Materials
Dibutyryladenosine-3’, 5’- cyclic monophosphate (DbcAMP), 6OHDA, ACEA (KD/I CB1 receptor 1.4 nM) [25] AM251 (KD/I CB1 receptor 7.49 nM), dihydroethidium (DHE) and LPS were used in this study; all reagents were from Sigma (St Louis, MO, USA). H2O2 was from Synth (Diadema, Brazil).
Cell Culture
A neuronal cell line derived from a mouse neuroblastoma (Neuro2A; American Type Culture Collection, Richmond, VA, USA) were plated (7x105 cells/mL) in 75 mm culture bottles in Dulbecco’s Modified Eagle Medium (DMEM) (Cultilab, Campinas, Brazil) supplemented with 10% fetal bovine serum (FBS), 3.9 mM L-glutamine (Gibco, Waltham, MA, USA) and 25 mM glucose. Cells were maintained at 37°C, in an atmosphere of 5% CO2 and saturated humidity. Cell differentiation was performed as previously described [26]. Briefly, the medium was replaced by DMEM with 0.5% FBS plus 1 mM DbcAMP. Dopaminergic phenotype was confirmed after 2 and 3 days by the increase of tyrosine hydroxylase (TH) expression. By HPLC dopamine quantification, Tremblay et al. [26] confirmed that 3 days of DbcAMP appropriately differentiate Neuro2A cells into dopaminergic-like cells.
Treatments
After 3 days of differentiation, cells were exposed to 6OHDA (50 μM) [27], LPS (30 μg/mL) [28], H2O2 (40 μM) [29], ACEA (1 and 2 μM) and/or AM251 (1 μM) for 24 hours. The doses of the injury treatments were experimentally determined to set cell death in 30%. Previous evidences proposed a dual effect of cannabinoids [30–32]. Additionally, ACEA presents a pattern of dose-related cytotoxicity. Low (in the order of 0.5 μM) and high (in the order of 10 μM) doses induce cell death [25,33]. In this regard, we first evaluate the ACEA (and AM251) cytotoxic pattern in our experimental model through a dose-response curve, in terms of cell viability (Figure 1B, 1C). ACEA and AM251 presented the same inverted U-shaped cell viability previously published [34], consequently, intermediate doses (ACEA 1 and 2 μM, AM251 1 μM) were chosen in order to avoid cannabinoid cytotoxicity falsepositive results [34]. AM251, a well known CB1 antagonist, was co-administered with ACEA in order to evaluate the cannabinoid receptor involvement during ACEA neuroprotective effect. The AM251 administration alone did not prevent cell death in each injury models (data not shown). The cell viability was evaluated by thiazoyl blue tetrazolium bromide (MTT) colorimetric assay. To evaluate the effect of ACEA and AM251 in differentiated Neuro2A cells, dose-response curves were done by measuring cell viability by the MTT method described below. The differentiated Neuro2A cell were divided in experimental groups as follows: control (Dimethyl sulfoxide, DMSO, treated), injury group (6OHDA, LPS and H2O2), injury and ACEA 1 and 2 μM treated cells and injury and ACEA/AM215 (1:1 and 2:1) co-treatment.
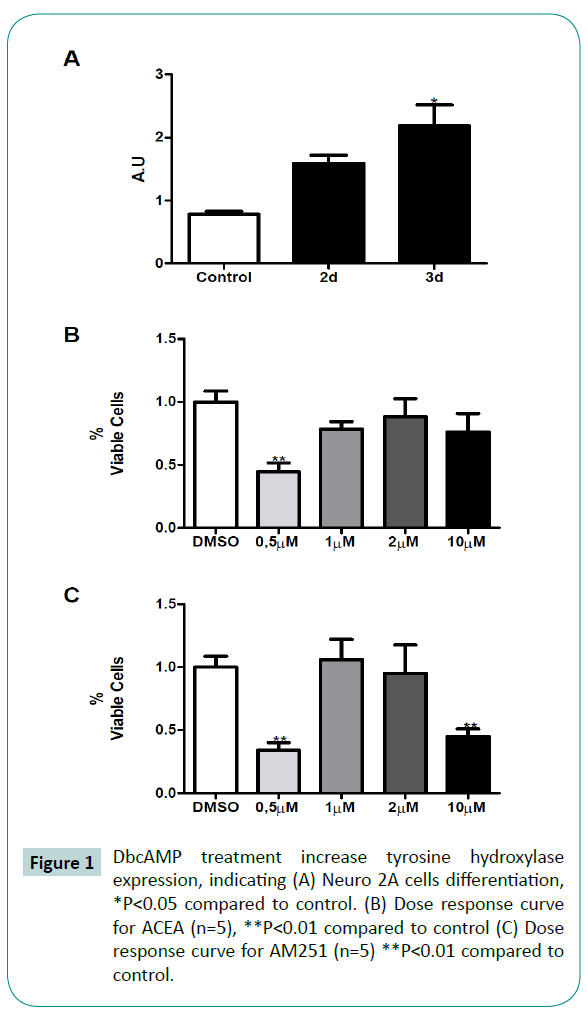
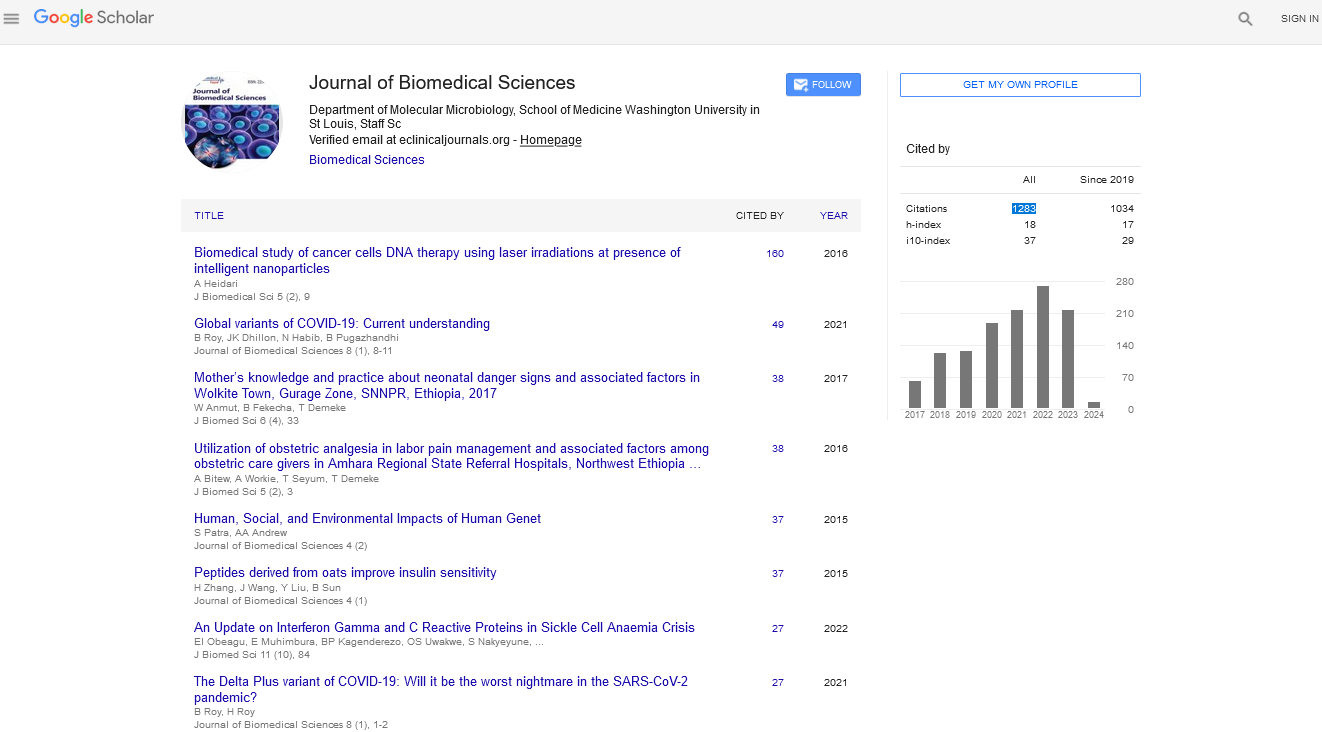
Figure 1: DbcAMP treatment increase tyrosine hydroxylase expression, indicating (A) Neuro 2A cells differentiation, *P<0.05 compared to control. (B) Dose response curve for ACEA (n=5), **P<0.01 compared to control (C) Dose response curve for AM251 (n=5) **P<0.01 compared to control.
Cell viability
After 24 h of treatments, cell viability was analyzed using a MTT reduction assay kit (Amresco, Solon, OH, USA) according to the manufacture instructions. Briefly, cells were incubated in a 5 mg/mL MTT solution in Phosphate-buffer saline (PBS) (concentrations in mM: NaCl 137, KCl 2.7, Na2HPO4 10, KH2PO4 1.8, pH 7.4) diluted (1:10) in serum-free DMEM medium for 2.5 h at 37°C. Supernatant was discarded and the cells solubilized in DMSO. The absorption values were determined at 550 nm in a microtiter plate reader (BioTek, Winooski, VT, USA). Cell viability is expressed as percentage of control (DMSO treated) cells.
ROS assay
Reactive oxygen species were analyzed in order to evaluate the oxidative cell damage. To this aim, dihydroethidium (DHE) was used as previously described [35] with few modifications. Briefly, cells were washed twice with ice cold PBS and incubated 10 min with 50 μMDHE diluted in serum-free DMEM. Supernatant was discarded and cells were trypsinized and centrifuged at 2,000 g for 5 min. Cell pellets were resuspended in Ca2+-free solution (concentrations in mM: NaCl 138, KCl 5.6, MgCl2 1.2, EGTA 1, HEPES 5, glucose 3, BSA 0.1 g, pH 7.4) and transfer to 96-well plates through 22 μm filters (BD Biosciences, East Rutherford, NJ, USA). Fluorescence was measured by flow-cytometry at 670 nm in a Guava EasyCyte TM Ht Sampling cytometer (Millipore, Darmstadt, Germany). ROS production is expressed as the percentage of mean fluorescence intensity of control (DMSO treated) cells.
Western blot
After 24 h of treatments cells were collected in Tris-buffered saline lysis buffer (TBS) containing 10 % NP-40, 10 % glycerol, 1 mM EDTA, 1 mM EGTA, 100 mM sodium orthovanadate, 1 M sodium pyrophosphate, 2 mM sodium fluoride and complete Protease inhibitor (Roche, Basel, Switzerland)36, sonicated and centrifuged at 15,000 g for 15 min in a Eppendorf 5480R centrifuge (Hamburg, Germany). Samples containing 30 μg of protein diluted in 100 mM DTT-supplemented Laemli buffer were resolved in 10% SDS-PAGE and electrotransferred to nitrocellulose membranes (0.22 μm ) using a Trans-Blot cell system (Bio-Rad). Membranes were incubated with anti-caspase 3 antibodies (1:1,000) (Cell Signaling, Beverly, MD, USA). Caspase 3 is a well-known apoptosis marker; increases in its expression is used as a marker of apoptosis-dependent cell death. Caspase 3 expression after 6OHDA treatment was analyzed by a timecourse experiment considering 10, 15 and 20 min time points after 6OHDA treatment (we present only the 20 min result). Tyrosine hydroxylase was detected by incubating membranes with anti TH antibody (1:1,000). After secondary antibody (horseradish peroxidase-coupled) incubation (1:10,000), proteins were detected by chemiluminescent method. Loading controls were performed with anti-μ-actin antibody. Western blots were quantified as band optical density by ImageJ Software (Wayne Rasband, NIH, Bethesda, MD, USA). Arbitrary units, A.U, are defined as the protein band optical density divided by the loading control band optical density.
Statistical analysis
One-way ANOVA with Tukey’s post hoc test was used for cellular viability analysis, ROS assay and western blot protein quantification. Statistical significance was set at P<0.05.
Results
ACEA and AM251 independent treatments does not induce cell death after 24 h treatment in DbcAMP differentiated Neuro2A cells.
In order to evaluate the protective role of cannabinoids in dopaminergic cells against well-known neuronal injuries, we first differentiated Neuro2A cells by 3-days treatment with DbcAMP and evaluate the ACEA and AM251 effect in cellular viability. After 3 days DbcAMP treatment we observed a significant increase in TH expression by means of one-way ANOVA analysis (Figure 1A), indicating dopaminergic-like phenotype, in agreement with previously published results [36]. The 24 h treatment with increasing doses of cannabinoids shown that 1 and 2 μM of ACEA did not induce cell death, neither 1 μM of AM251 (Figure 1B and 1C). With these results, we rule out the possibility that cannabinoids are contributing to cell death in our model. Consequently, we chose those cannabinoids concentrations to evaluate their protective effect against 6OHDA, LPS and H2O2.
CB1 receptor agonist ACEA increases dopaminergic cell viability after neurotoxic, inflammatory and oxidative processes.
Exposure of differentiated Neuro 2A cells to 6OHDA (50 μM), LPS (30 μg/mL), and H2O2 (40 μM), significantly reduced cellular viability in 30%, 31% and 38%, respectively, compared to control (Figure 2A-C, second bar). The 1 μM ACEA treatment did not significantly prevent the cytotoxic challenge after 6OHDA (P=0.0094; F=3.298), LPS (P<0.0001; F=7.421) and H2O2 (p<0.0001; F=11.28) (Figure 2A-C, third bar). However, 2 μM of ACEA treatment was able to prevent, at least partially, the cells from death induced by 6OHDA (12% cell death), LPS (10% cell death) or H2O2 (7% cell death).
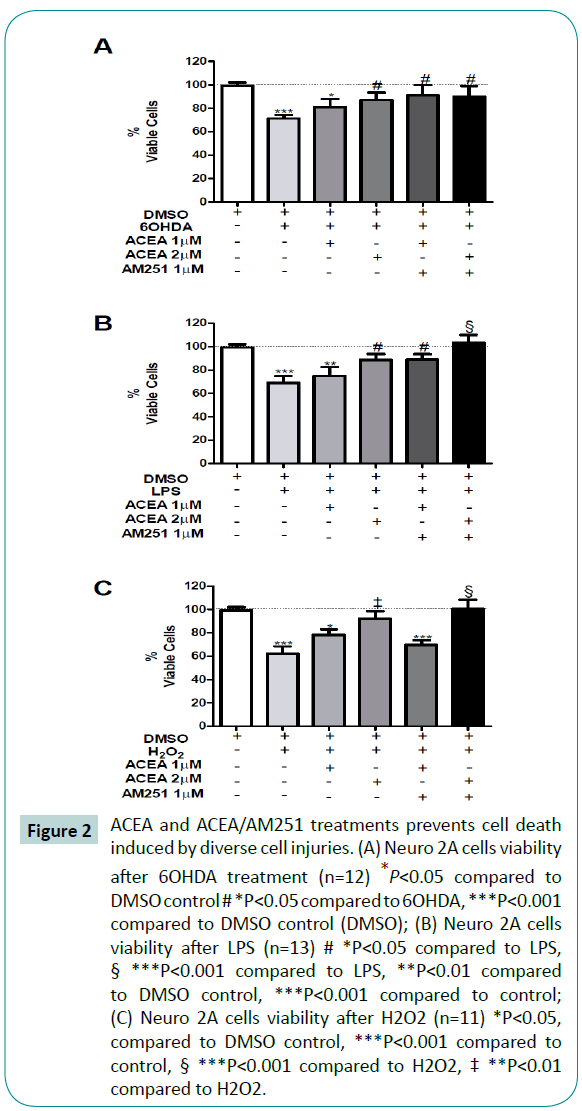
Figure 2: ACEA and ACEA/AM251 treatments prevents cell death induced by diverse cell injuries. (A) Neuro 2A cells viability after 6OHDA treatment (n=12) *P<0.05 compared to DMSO control # *P<0.05 compared to 6OHDA, ***P<0.001 compared to DMSO control (DMSO); (B) Neuro 2A cells viability after LPS (n=13) # *P<0.05 compared to LPS, § ***P<0.001 compared to LPS, **P<0.01 compared to DMSO control, ***P<0.001 compared to control; (C) Neuro 2A cells viability after H2O2 (n=11) *P<0.05, compared to DMSO control, ***P<0.001 compared to control, § ***P<0.001 compared to H2O2, ‡ **P<0.01 compared to H2O2.
ACEA is a well-known CB1 receptor-specific agonist [25]. To evaluate whether the ACEA protective effect succeeded in activating CB1 receptor, we challenged the cells with a CB1 receptor antagonist, AM251. Surprisingly, the administration of ACEA + AM251 in both, 1:1 (Figure 2A-C, fifth bar), but H2O2, and 2:1 stoichiometry in all treatments, did not reverse the protective effect of ACEA (Figure 2A-C, sixth bar). Thus, the ACEA and AM251 combined treatment (2:1) blocked the LPS and H2O2 cytotoxic effect, however, in the presence of 6OHDA, the antagonist AM251 did not present either effect.
CB1 receptor agonist ACEA protects dopaminergic cell from inflammatory and oxidative-induced death by decreasing ROS production.
To evaluate the antioxidant function of cannabinoids with the cell death prevention previously seen (Figure 2) we analyze the ROS production during the same treatments. In agreement with the previous cellular viability results, the exposure of differentiated Neuro 2A cells to 6OHDA, LPS and H2O2, significantly increased ROS production in 52%, 51% and 159%, respectively, compared to control (Figure 3) with a concomitant reduction in cellular viability of 30%, 31% and 38% respectively (Figure 2). Despite the protective role of ACEA (2 μM) against 6OHDA, we did not observe a significant reduction in ROS production (P=0.0043; F=7.941) (Figure 3A). On the other hand, the 2 μM ACEA co-treatment with 1 μM AM251 completely restores the ROS production to the control levels after 6OHDA and LPS exposure, respectively (Figure 3A and 3B).
Figure 3: ACEA and ACEA/AM251 (2:1) reduces reactive oxygen species production in Neuro 2A cells. (A) ACEA/AM251 2:1 reduce ROS production after 24 h 6OHDA treatment (n=6) **P<0.01 compared to control, #*P<0.05 compared to 6OHDA; (B) LPS-induced ROS production reduction after ACEA/AM251 2:1 treatment (n=6) **P<0.01 compared to control, # *P<0.05 compared to LPS; (C) Neuro 2A ROS production after H2O2 (n=6) **P<0.01 compared to control, # *P<0.05 compared to H2O2,† *P<0.05 compared to DMSO+H2O2+ACEA 1 μM.
In order to evaluate the participation of CB1 receptor in this presumptive ACEA-mediated antioxidant effect, the coadministration of AM251 was conducted. The administration of ACEA + AM251 was not able to reverse the antioxidant effects of ACEA against LPS (Figure 3B) but completely reverse it against H2O2 (Figure 3C). These results suggest that cannabinoids exert a protective role against cellular injuries, in part by reducing the oxidative species.
The protective role of cannabinoids is partially due to the reduction in apoptosis-mediated cell death
Caspase 3 is a well-known apoptosis marker. To evaluate if the contribution of cannabinoids in cell survival was due to an apoptosis-reduction, we analyzed the caspase 3 expression. As expected, 6OHDA (Figure 4A), LPS (Figure 4B) and H2O2 (Figure 4C) increased caspase 3 expression. When cells were treated with ACEA, a significant reduction in caspase 3 expression was observed in cells exposed to 6OHDA, LPS and H2O2. When cotreated with ACEA and AM251 (1:1), we observed a reduction tendency in caspase 3 expression during 6OHDA and H2O2 injuries (Figure 4A and 4B), however, not statistically significant; ACEA/ AM251 significantly reduced caspase 3 expression during LPS injury. These results suggest that the neuroprotective effect of cannabinoids is partially due to a reduction in the apoptosismediated cell death, by means of caspase-3 expression reduction.
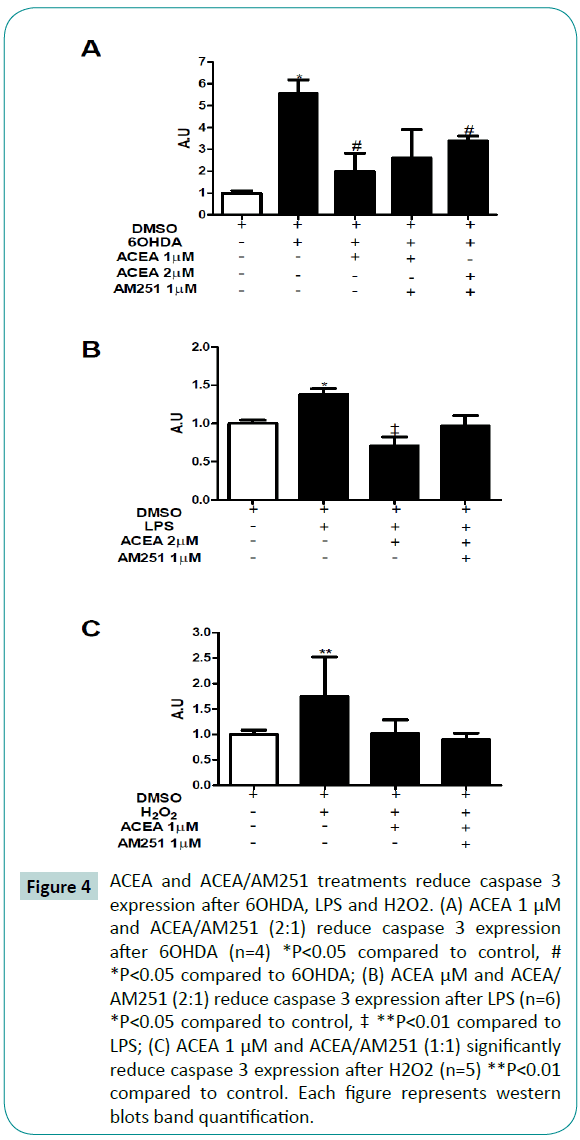
Figure 4: ACEA and ACEA/AM251 treatments reduce caspase 3 expression after 6OHDA, LPS and H2O2. (A) ACEA 1 μM and ACEA/AM251 (2:1) reduce caspase 3 expression after 6OHDA (n=4) *P<0.05 compared to control, # *P<0.05 compared to 6OHDA; (B) ACEA μM and ACEA/ AM251 (2:1) reduce caspase 3 expression after LPS (n=6) *P<0.05 compared to control, ‡ **P<0.01 compared to LPS; (C) ACEA 1 μM and ACEA/AM251 (1:1) significantly reduce caspase 3 expression after H2O2 (n=5) **P<0.01 compared to control. Each figure represents western blots band quantification.
Discussion
In this study, we show that cannabinoid compounds ACEA and AM251 prevent dopaminergic neurons death induced by neurotoxic, inflammatory and oxidative stimuli in vitro. This neuroprotective effect was associated to a reduction in ROS production and caspase 3 expression.
Endocannabinoid system has been implicated in neurodegenerative processes and neuroprotective properties of cannabinoids have been suggested. Part of this protection seems to occur by the activation of cannabinoid receptors or through an antioxidant capacity of cannabinoids [4,6,7,12-20,37].
ACEA is a well-known cannabinoid receptor agonist widely use in cannabinoid system research. However, there are controversial data in the literature about the cytotoxicity of CB1 receptor activation. In this regard, Caltana et al. [33] recently demonstrated a dose-dependent relation within ACEA effect, where low doses did not induce cell death by means of increase in Caspase 3 positive cells. Dose-response curves analysis showed that low doses ACEA increase cell death, however, higher ACEA concentration prevent cell death (Figure 1B). This result agrees with previous works that states the typical inverted U-shaped dose-response curve of ACEA and cell viability [38]. Our data suggest that 1 and 2 μM ACEA did not induce statistically significant cell death in our model (Figure 1B). In this regard, we used these concentration during this study for avoid possible artifacts during oxidative and inflammatory cell injury models.
Cannabinoids have been proposed as promising drugs for neurodegenerative treatment [2,31,39]. In order to evaluate a neuroprotective effect of ACEA in in vitro dopaminergic-like differenced Neuro2A cells, we evaluated cellular viability after cytotoxic events. During Parkinson’s disease occurs a massive loss of dopaminergic neurons at the susbstancia nigra [23,40– 44]. It has been proposed dopaminergic-induced cytotoxicity and inflammatory processes as the events responsible for neuronal loss, both related with oxidative damage. In order to mimic oxidative and inflammatory neurotoxicity, we used 6OHDA, LPS and H2O2, all previously set at a concentration that induce 30% cell death (Figure 2A-C, light gray bar). When treated with ACEA 1 μM, we observed a reduction in cell death to 20% after 6OHDA, 26% after LPS and 22% after H2O2 (Figure 2A-C third bar). By doubling ACEA concentration, we observed cell death reduction to 13% with 6OHDA, 12% with LPS and 8% with H2O2 (Figure 2A-C fourth bar), that represents a significant recovery compared with injury treatment alone. These data suggest a protective role of ACEA against oxidative and inflammatory damages of dopaminergiclike neurons. ACEA presents high affinity for CB1 receptor (1.4 nM). However, our cellular viability data do not provide direct evidence about the involvement of the cannabinoid system in dopaminergic-like differenced Neuro2A cells.
In order to evaluate the cannabinoid receptor-mediated cell survival, we used AM251, a well-known CB1 receptor antagonist; AM251 presents an inhibition constant of 7.49nM. During 6OHDA and LPS treatment, AM251 did not blocked the ACEA protective effect (Figure 2A and 2B). As demonstrated in another experimental model, Baker and McDougall [45] have also observed the same effect. Briefly, the use of AM251 did not modify the ACEA-dependent increase in blood perfusion in rat knee joint. In this regard, the absence of an increase in cell death because of the AM251-mediated blockade of ACEA protection effect opens the possibility to the participation of receptors other than CB1. By using capsazepin, transient receptor potential channel vanilloid receptor 1 (TRPV1) antagonist, Baker and McDougall [45] shown an impairment in ACEA effect. Preliminary results of our group suggest capsazepin effect in neuronal viability. Capsazepin and ACEA co-treatment resulted in an increase in cell death against 6OHDA (data not shown). Since anandamide and ACEA activates CB1 and TRPV1, the vanilloid receptor could be associated to the cannabinoid system [46,47]. In this sense, our results may suggest that the ACEA neuroprotection observed could be related to the cannabinoid system, presumably by means of CB1 and TRPV1 activation. It should be noted that cannabinoid system does not functions in a canonical way, since ACEA activates CB1 so as TRPV1 receptors.
The increase in oxygen reactive species during Parkinson’s disease is well documented [42,48]. Recent data suggests the interaction between oxidative stress and inflammation that in vivo contributes to dopaminergic cell death [49]. Several endogenous antioxidant mechanisms have been studied [50] from which the cannabinoids rise up as a new promise against oxidative damage [51]. To understand the mechanism related to the ACEAdependent neuroprotection previously shown, we evaluated the ROS production. All cellular injury models increase cell death (Figure 2) with a concomitant rise in ROS production (Figure 3). Interestingly, the 30% cell death induced by 6OHDA, LPS and H2O2 (H2O2 increased 38%, Figure 2C) led to a substantial increase in ROS compared to control (52% by 6OHDA, 51% by LPS and 159% by H2O2). These results suggest that neuronal cell death is partially due to oxidative damage. The 2 μM ACEA reduced ROS production after 6OHDA and LPS; 1μM ACEA significantly reduced H2O2-induced ROS production (Figure 3A-C third bar). Previous evidences have established that cannabinoids has receptor independent antioxidant potential [52], presumably by increasing mRNA levels for Cu, Zn-Superoxide dismutase 5. In this regard, considering the significant reduction in ROS production, we do not rule out the possibility of an additional ACEA antioxidant mechanism, independent of CB1/TRPV1 receptors.
To evaluate cannabinoid receptor CB1 involvement during ROS production inhibition, we co-administrated AM251, selective CB1 antagonist [53]. According with the cell viability result, AM251 do not inhibit the ACEA-mediated ROS reduction during 6OHDA and LPS injuries (Figure 3A, 3B, fourth bar). In this sense, the ACEA-dependent ROS reduction may be due to the activation of TRPV1 receptors. However, AM251 was capable to inhibit the ACEA-dependent protection during H2O2 (Figure 3C, fourth bar). The endocannabinoid system has been proposed to be critical for defining the cellular fate, which varies from each endocannabinoid, cellular injury and environment [31,54,55]. This result may suggest that during a cytotoxic event induced by oxidant species, the cannabinoid system function mainly through cannabinoid receptors, i.e. CB1, more than through TRPV1, explaining the AM251 mediated rise in ROS production.
All three-injury models presented in this work induce cell death with a concomitant increase in caspase 3 expression. In this regard, we evaluated if the ACEA-dependent cell survival was due to a reduction in caspase 3 expression, well-known apoptosis marker. Interestingly, other cell death modalities, like autophagy, has been described to be related with Parkinson’s disease in neuroblastoma cell model [56]. All the injury models, set at 30% cell death, induce an increase in more than 50% of ROS production (Figure 3A-C, second bar) with a concomitant increase in caspase 3 expression (Figure 4A-C, second bar). In agreement with cell survival and ROS production data, an ACEA-mediated significant reduction in caspase 3 expression was observed (Figure 4A-C, third bar). Additionally, AM251 co-treatment did not increase caspase 3 expression because of an ACEA blockade effect. Considering that caspase 3 expression is the ultimate event previous to cell death, it is necessary the activation of intracellular pathways that result in apoptosis. Since the ACEA administration leads to a reduction in caspase 3 expression, it is possible that the cannabinoid exerted a regulatory effect in key intracellular pathways in our model.
Numerous proteins have been associated in the control of cell death/survival decision. Extracellular signal-regulated kinases 1 and 2 (ERK 1/2) are members of the large serine/threonine family of mitogen-activated protein kinases (MAPKs) which have been related to several cellular functions, including proliferation, differentiation and neuronal death/survival [54,57]. Growing number of evidences suggest a death-promoting role for ERK 1/2 in distinct models of neuronal death [58]. Additionally, ERK 1/2 is activated by ROS, 6OHDA and G coupled receptors [59,60]. However, it has been suggested that ERK pathway activation may be compromised with both cell death and survival, having a dual role in cell fate [61,62]. In this regard, cannabinoids may control the cell fate by modulating ERK pathway depending in the level of cellular damage because of environmental cytotoxicity. It should be noted that the cannabinoid system does not functions in a canonical fashion. Endocannabinoids presents retrograde signaling and systemically ubiquitous receptor localization. CB1 and CB2 activation by endocannabinoids have been associated with more than a unique response, having opposite effects CB1 activation in peripheral tissues and in the central nervous system.
It should be noted that the absence of inhibitory effect of AM251 during cell viability, ROS production and caspase 3 expression experiments is not a definitive parameter for establishing that the ACEA protective effects are not dependent of cannabinoid receptor activation. More experiments should be done for clearly identify the binding of ACEA to CB receptors in our model. However, because of the high affinity of ACEA for CB1 receptor ( Ki= 1.4 nM25), we could not rule out the possibility of the involvement of cannabinoid pathways activation via CB receptors. In the same time, it has been proposed that AM251 may exert allosteric modulation of cannabinoid receptor CB1 [44-46], opening the possibility for a wide range of effects in the receptor activity.
In conclusion, CB1 receptor agonist ACEA protects dopaminergic neurons from neurotoxicity, inflammation and oxidative stress in a partial cannabinoid receptor-dependent manner. These neuroprotection seems to occur by reducing ROS production and caspase 3 activation during neurotoxic, inflammatory and oxidative events.
Competing of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported in this paper.
Author contribution
Batinga H carried out experimental design, experimental procedures, data analysis and paper writing. Zuniga-Hertz JP carried out experimental procedures (western blot) and paper writing. Torrao AS carried out experimental design, paper writing and reviewing.
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 2011/51971-0 and 2014/06372- 0). Batinga H. was the recipient of fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 140170/2012-0) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 2013/02916-2).
Acknowledgment
We acknowledge MSc. Caio Mazucanti (University of Sao Paulo) for critical comments and his technical support during the process. We thank to professors Dr. Luiz Britto, Dr. Cristoforo Scavone and Dr. Angelo Carpinelli (University of Sao Paulo) for their laboratory facilities.
7662
References
- Marzo V Di (2006)A brief history of cannabinoid and endocannabinoid pharmacology as inspired by the work of British scientists. Trends Pharmacol Sci 27:134–140.
- Youssef FF, Irving AJ (2012)From cannabis to the endocannabinoid system: refocussing attention on potential clinical benefits. West Indian Med J 61:264–270.
- Ameri A (1999)The effects of cannabinoids on the brain.Prog. Neurobiol 58:315–348.
- Stelt M van der, Marzo V Di (2005) Cannabinoid receptors and their role in neuroprotection. Neuromolecular Med 7:37–50.
- García-arencibia M, González S, Lago E de, Ramos J a, Mechoulam R, et al. (2007) Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res 1134:162–170.
- Lago E de, Fernández-Ruiz J (2007) Cannabinoids and neuroprotection in motor-related disorders. CNS Neurol. Disord. Drug Targets6:377–387.
- Sánchez AJ, García-Merino A (2012)Neuroprotective agents: cannabinoids. Clin. Immunol142:57–67.
- Giacoppo S, Mandolino G, Galuppo M, Bramanti P, Mazzon E (2014) Cannabinoids: new promising agents in the treatment of neurological diseases. Molecules 19:18781–18816.
- Casteels C, Lauwers E, Baitar A, Bormans G, Baekelandt V, et al. (2010) In vivo type 1 cannabinoid receptor mapping in the 6-hydroxydopamine lesion rat model of Parkinson’s disease. Brain Res 1316:153–162.
- González S, Scorticati C, García-Arencibia M, Miguel R de, Ramos JA, et al. (2006) Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson’s disease. Brain Res 1073-1074:209–219.
- Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, et al. (2001)Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem 78:1415–1427.
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, et al. (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature413:527–531.
- Unzicker C, Erberich H, Moldrich G, Woldt H, Bulla J, et al. (2005) Hippocampal cannabinoid-1 receptor upregulation upon endothelin-B receptor deficiency: a neuroprotective substitution effect? Neurochem Res 30:1305–1309.
- Walsh S, Mnich K, Mackie K, Gorman AM, Finn DP, et al. (2010) Loss of cannabinoid CB 1 receptor expression in the 6-hydroxydopamine-induced nigrostriatal terminal lesion model of Parkinson ’s disease in the rat. Changes. 81:543–548.
- Romero J, Berrendero F, Pérez-Rosado A, Manzanares J, et al. (2000)Unilateral 6-hydroxydopamine lesions of nigrostriatal dopaminergic neurons increased CB1 receptor mRNA levels in the caudate-putamen. Life Sci 66:485–494.
- Stelt M van der, Veldhuis WB, Haaften GW van, Fezza F, Bisogno T, et al. (2001)Exogenous anandamide protects rat brain against acute neuronal injury in vivo. JNeurosci 21:8765–8771.
- Glass M, Dragunow M, Faull RL (2000) The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience 97:505–519.
- Laere K Van, Casteels C, Lunskens S, Goffin K, Grachev ID, et al. (2012) Regional changes in type 1 cannabinoid receptor availability in Parkinson’s disease in vivo. Neurobiol Aging33:620.e1–8.
- Grundy RI, Rabuffetti M, Beltramo M (2001) Cannabinoids and neuroprotection. MolNeurobiol24:29–51.
- Sagredo O, García-Arencibia M, Lago E de, Finetti S, DecioA,et al. (2007) Cannabinoids and neuroprotection in basal ganglia disorders. MolNeurobiol36:82–91.
- Stelt M Van der, Marzo V Di, Stelt M Van Der, Marzo V Di (2003)The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: implications for neurological and psychiatric disorders. Eur J Pharmacol 480:133–150.
- Romero J, Lastres-Becker I, Miguel R de, Berrendero F, Ramos JA (2002) The endogenous cannabinoid system and the basal ganglia. biochemical, pharmacological, and therapeutic aspects. PharmacolTher95:137–152.
- Lastres-Becker I, Molina-holgado F, Ramos AJ,Mechoulam R, FernaJ,et al.(2005) Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro?: Relevance to Parkinson ’ s disease.Neurobiol Disease 19:96 – 107.
- Chaves-Kirsten GP, Mazucanti CHY, Real CC, Souza BM, Britto LRG, et al. (2013)Temporal changes of CB1 cannabinoid receptor in the basal ganglia as a possible structure-specific plasticity process in 6-OHDA lesioned rats. PLoS One 8:e76874.
- Hillard CJ, Manna S, Greenberg MJ, Dicamelli R, Ross RA, et al. (1999) Synthesis and Characterization of Potent and Selective Agonists of the Neuronal Cannabinoid Receptor ( CB1 ) 1. J PharmacolExpTher289:1427–1433.
- Tremblay RG, Sikorska M, Sandhu JK, Lanthier P, Ribecco-Lutkiewicz M, et al. (2010) Differentiation of mouse Neuro 2A cells into dopamine neurons. J Neurosci Methods 186:60–67.
- Lundius EG, Stroth N, Vukojevi? V, Terenius L, Svenningsson P (2013) Functional GPR37 trafficking protects against toxicity induced by 6-OHDA, MPP+ or rotenone in a catecholaminergic cell line. JNeurochem 124:410–407.
- Rodríguez-González R, Baluja A, Veiras Del Río S, Rodríguez A, Rodríguez J, et al. Effects of sevofluranepostconditioning on cell death, inflammation and TLR expression in human endothelial cells exposed to LPS. J Transl Med 11:87.
- Calderón FH, Bonnefont A, Muñoz FJ, Fernández V, Videla LA, et al. (1999) PC12 and neuro 2a cells have different susceptibilities to acetylcholinesterase-amyloid complexes, amyloid25-35 fragment, glutamate, and hydrogen peroxide. J Neurosci Res 56:620–631.
- Sarne Y, Mechoulam R (2005) Cannabinoids: between neuroprotection and neurotoxicity. Curr. Drug Targets. CNS NeurolDisord 4:677–684.
- Sarne Y, AsafF, Fishbein M, Gafni M, Keren O (2011)The dual neuroprotective-neurotoxic profile of cannabinoid drugs. Br JPharmacol 163:1391–1401.
- Sarne Y, Keren O (2004) Are cannabinoid drugs neurotoxic or neuroprotective? Med. Hypotheses 63:187–192.
- Caltana LR, Heimrich B, BruscoA (2015) Further Evidence for the Neuroplastic Role of Cannabinoids: A Study in Organotypic Hippocampal Slice Cultures. J MolNeurosci 56:773–781.
- Nazario LR, Antonioli R, Capiotti KM, Hallak JEC, Zuardi AW, et al. (2015) Caffeine protects against memory loss induced by high and non-anxiolytic dose of cannabidiol in adult zebrafish (Daniorerio). PharmacolBiochemBehav 135:210–216.
- Lucena CF, Roma LP, Graciano MFR, Veras K, Simões D, et al. (2015) Omega-3 Supplementation Improves Pancreatic Islet Redox Status. Pancreas 44:287–295.
- Molina-holgado E, Vela M, Arévalo-Martín A, Molina-Holgado F, Borrell J, et al.(2002) Cannabinoids Promote Oligodendrocyte Progenitor Survival?: Involvement of Cannabinoid Receptors and Phosphatidylinositol-3 Kinase / Akt Signaling. JNeurosci22:9742–9753.
- Bisogno T, Marzo V Di (2008)The role of the endocannabinoid system in Alzheimer’s disease: facts and hypotheses. Curr Pharm Des 14:2299–3305.
- Koch JE, Matthews SM (2001) Delta9-tetrahydrocannabinol stimulates palatable food intake in Lewis rats: effects of peripheral and central administration. Nutr. Neurosci. 4:179–187.
- Tucci SA, Halford JCG, Harrold JA, Kirkham TC (2006)Therapeutic potential of targeting the endocannabinoids: implications for the treatment of obesity, metabolic syndrome, drug abuse and smoking cessation. Curr MedChem 13:2669–2680.
- Ferrari CC, TarelliR (2011) Parkinson’s disease and systemic inflammation. Parkinsons Dis1–9.
- Hald A, Lotharius J (2005) Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? ExpNeurol193:279–290.
- Whitton PS (2007) Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br JPharmacol150:963–976.
- Brotchie JM (2003) CB 1 cannabinoid receptor signalling in Parkinson ’ s disease. CurrOpinPharmacol 3:54–61.
- Machado a, Herrera a J, Venero JL, Santiago M, Pablos RM De, et al. (2011) Peripheral inflammation increases the damage in animal models of nigrostriatal dopaminergic neurodegeneration: possible implication in Parkinson’s disease incidence. Parkinsons Dis 2011:393769.
- Baker CL, McDougall JJ (2004)Thecannabinomimetic arachidonyl-2-chloroethylamide (ACEA) acts on capsaicin-sensitive TRPV1 receptors but not cannabinoid receptors in rat joints. Br J Pharmacol142:1361–1367.
- Lisboa SF, Guimarães FS (2012) Differential role of CB1 and TRPV1 receptors on anandamide modulation of defensive responses induced by nitric oxide in the dorsolateral periaqueductal gray. Neuropharmacology 62:2455–2462.
- Marzo V Di, Petrocellis L De (2012)Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R SocLond B BiolSci367:3216–3228.
- Farooqui T, Farooqui A a(2011) Lipid-mediated oxidative stress and inflammation in the pathogenesis of Parkinson’s disease. Parkinsons Dis 1–9.
- Sharma N, Nehru B (2015) Characterization of the lipopolysaccharide induced model of Parkinson’s disease: Role of oxidative stress and neuroinflammation. NeurochemInt 87:92–105.
- Hampson AJ,Grimaldi M, Axelrod JA, Wink D (1998) Cannabidiol and (-) DELTA 9-tetrahydrocannabinol are neuroprotective antioxidants. ProcNatlAcadSciUSA 95:8268–8273.
- Harvey BS, Ohlsson KS, Mååg JL V, Musgrave IF, Smid SD (2012) Contrasting protective effects of cannabinoids against oxidative stress and amyloid-β evoked neurotoxicity in vitro. Neurotoxicology33:138–146.
- Hampson AJH, Grimaldi MG, Axelrod JA, Wink D (1998)Cannabidiol and (-)delta 9-tetrahydrocannabinol are neuroprotective antioxidants. Med Sci 95:8268–8273.
- Gatley SJ, Gifford a N, Volkow ND, Lan R, Makriyannis(1996) a. 123I-labeled AM251: a radioiodinated ligand which binds in vivo to mouse brain cannabinoid CB1 receptors.Eur J Pharmacol 307:331–338.
- Turu G, Hunyady L (2010) Signal transduction of the CB1 cannabinoid receptor. JMol Endocrinol44:75–85.
- Fowler CJ, Rojo ML, Rodriguez-gaztelumendi A (2010) Modulation of the endocannabinoid system?: Neuroprotection or neurotoxicity?? Exp Neurol 224:37–47.
- Chen Y, Wang S, Zhang L, Xie T, Song S, et al. (2015) Identification of ULK1 as a novel biomarker involved in miR-4487 and miR-595 regulation in neuroblastoma SH-SY5Y cell autophagy. SciRep 5:11035.
- Davis S, MirickDK(2006) Circadian disruption, shift work and the risk of cancer: a summary of the evidence and studies in Seattle. Cancer causes Control CCC17:539–545.
- Galve-Roperh I, Rueda D, Gómez del Pulgar T, Velasco G, Guzmán M (2002) Mechanism of extracellular signal-regulated kinase activation by the CB(1) cannabinoid receptor. MolPharmacol 62:1385–1392.
- Kulich SM, Chu CT (2003) Role of reactive oxygen species in extracellular signal-regulated protein kinase phosphorylation and 6-hydroxydopamine cytotoxicity. JBiosci 28:83–89.
- Subramaniam S, UnsickerK (2010) ERK and cell death: ERK1/2 in neuronal death. FEBS J 277:22–29.
- Bouaboula M, Poinot-Chazel C, Bourrié B, Canat X, Calandra B, et al. (1995) Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J 312:637–641.
- Subramaniam S, Unsicker K (2006) Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience138:1055–1065.