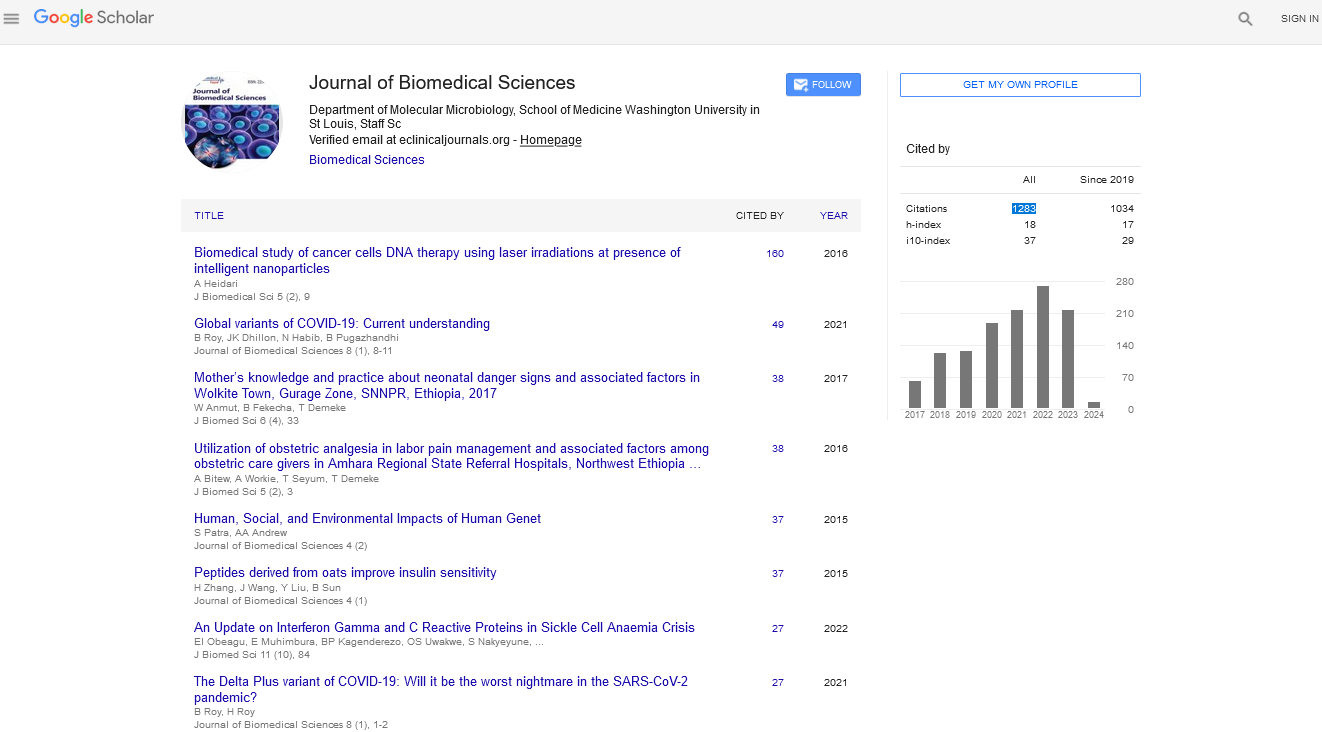
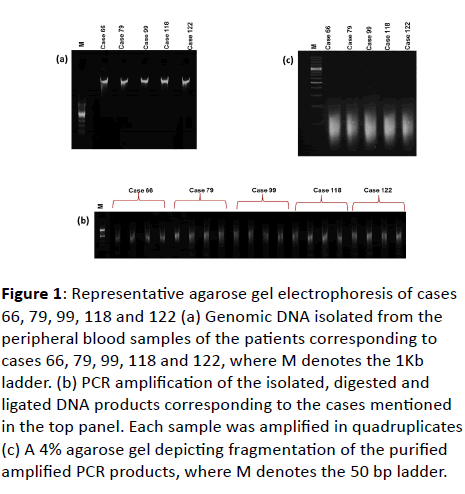
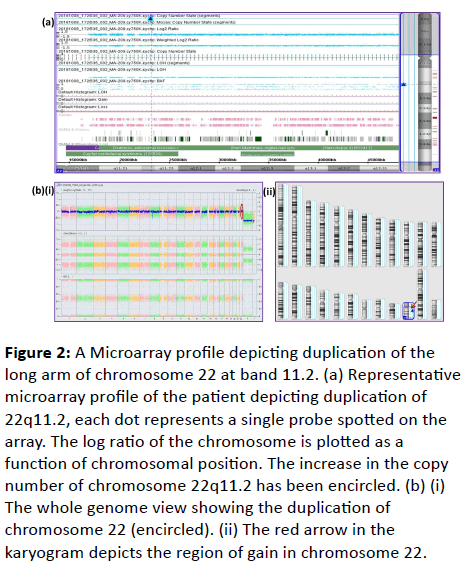
| 1 |
1 |
3Y/M |
Mild autism spectrum disorder, Speech delay, Poor eye contact, ADHD |
0 |
Loss |
1 |
q44 |
317 |
1 |
arr [hg19] 1q44 (246,174,603-246,491,736) × 1 |
Pathogenic |
| 2 |
3 |
2/F |
Autism spectrum disorder, Speech delay, Hyperactive, Slightly delayed milestones, Sensory issues |
3 |
Gain |
6 |
q11.1 |
1037 |
1 |
arr [hg19] 6q11.1(61,886,426-62,923,825) × 3 |
Pathogenic |
| 3 |
4 |
4/F |
Global developmental delay, facial dysmorphism |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 4 |
5 |
1Y/M |
Developmental delay with seizures |
3 |
Gain |
22 |
q11.22 |
484 |
2 |
arr [hg19] 22q11.22 (22,817,622-23,301,460) × 3 |
Pathogenic |
| 5 |
6 |
7Y/M |
ADHD with autism spectrum disorder |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 6 |
7 |
6Y/M |
Behavioural disorder, facial dysmorphism with ADHD |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 7 |
8 |
13Y/M |
Autism, Delayed milestones, Facial asymmetry, Supernumerary teeth, No eye contact |
1 |
Loss |
1 |
q21.1 |
124 |
2 |
arr [hg19] 1q21.1(144,950,048-145,074,541) ×1 |
Likely pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
330 |
1 |
arr [hg19] 22q11.22 (22,929,364-23,258,939) × 3 |
Likely pathogenic |
| 8 |
9 |
5Y/M |
Behavioural issues |
3 |
Gain |
10 |
q11.22 |
853 |
3 |
arr [hg19] 10q11.22 (46,293,590-47,147,021) × 3 |
Likely pathogenic |
| 9 |
10 |
3Y/M |
Developmental delay, Repetitive behaviour, Balance problem |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 10 |
11 |
2Y/F |
Gross motor delay, Hypotonia, Imbalance while walking, Dysmorphic facial features, Epicanthal folds |
3 |
Gain |
12 |
p13.31 |
500 |
7 |
arr [hg19] 12p13.31 (7,917,870-8,417,898) × 3 |
Likely pathogenic |
| 11 |
12 |
3Y/M |
Global developmental delay, ADHD |
3 |
Gain |
X |
p22.33 |
331 |
3 |
arr [hg19] Xp22.33 (313,456-644,440 or 263,456-94,440) × 3 |
Likely pathogenic |
| 12 |
13 |
10Y/M |
Weakness, ? Developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 13 |
14 |
4Y/M |
Dysmorphic facial features and developmental delay |
3 |
Gain |
13 |
q12.12 |
810 |
9 |
arr [hg19] 13q12.12q12.13 (25,446,646-25,736,308) × 3 |
Likely pathogenic |
| |
|
|
|
2 |
Gain |
X |
q27.1 |
609 |
1 |
arr [hg19] 13q12.12q12.13 (25,446,646-25,736,308) × 3 |
Likely pathogenic |
| 14 |
15 |
3Y/M |
Speech delay |
3 |
Gain |
22 |
q11.22 |
377 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 15 |
17 |
7M/M |
Multiple congenital anomalies, ASD?, B/L Retinoblastoma |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 16 |
18 |
33Y/F |
Risk of Down syndrome |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 17 |
20 |
6Y/F |
Failure to thrive, Speech delay, Gross motor delay, Intellectual disability, Increased Hypotonia, Seizures, Micrognathia, Retrognathia |
3 |
Gain |
22 |
q11.22 |
358 |
6 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 18 |
21 |
3Y/M |
Global Developmental Delay, ? fragile X syndrome, Intellectual disability |
3 |
Gain |
22 |
q11.22 |
358 |
6 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 19 |
22 |
11Y/F |
Behavioural issues, Speech Delay, Learning Disability, Intellectual Disability, Autism, Dimorphism |
3 |
Gain |
22 |
q11.22 |
377 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,278,835) × 3 |
Pathogenic |
| 20 |
23 |
4/F |
? Rett syndrome, Autism, Speech delay, Motor delay |
3 |
Gain |
22 |
q11.22 |
400 |
3 |
arr [hg19] 22q11.22 (22,901,370-23,301,460) × 3 |
Pathogenic |
| 21 |
28 |
2Y/F |
Developmental delay (motor and sensory) and stronger anxiety |
3 |
Gain |
22 |
q11.22 |
358 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 22 |
31 |
1 Y/M |
Global developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 23 |
32 |
1Y and 10 M/M |
Global developmental delay with hypotonia |
3 |
Gain |
22 |
q11.22 |
372 |
2 |
arr [hg19] 22q11.22 (22,929,364-23,301,460) × 3 |
Pathogenic |
| 24 |
33 |
9 Years/M |
Hyperactive, Seizures, Short hands and Hypogonadism |
3 |
Gain |
1 |
p36.33 |
67 |
1 |
arr [hg19] 1p36.33 (2,174,958-2,242,417) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
16 |
p13.3 |
160 |
8 |
arr[hg19] 16p13.3 (2,053,327-2,213,040) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
19 |
p13.3 |
937 |
35 |
arr [hg19] 19p13.3 (675,955-1,612,855) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
372 |
1 |
arr [hg19] 22q11.22 (22,929,364-23,301,460) × 3 |
Pathogenic |
| |
|
|
|
2 |
Gain |
X |
q13.1 |
62 |
3 |
arr [hg19] Xq13.1 (70,481,039-70,542,994) × 2 |
Pathogenic |
| 25 |
35 |
5 Y/M |
Autism Spectrum Disorder with ADHD |
1 |
Loss |
7 |
p21.3 |
172 |
1 |
arr [hg19] 7p21.3 (8,614,447-8,786,729) × 1 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
372 |
2 |
arr [hg19] 22q11.22 (22,929,364-23,301,460) × 3 |
Pathogenic |
| 26 |
36 |
4Y/F |
Behavioural issues, dysplastic toes and developmental delay |
3 |
Gain |
10 |
q11.22 |
3 |
|
arr [hg19] 10q11.22 (46,293,590-48,220,168) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
2 |
|
arr [hg19] 22q11.22 (22,901,370-23,259,033) × 3 |
Pathogenic |
| |
|
|
|
4 |
Gain |
X |
Q26.2 |
1 |
|
arr [hg19] 22q11.22(22,901,370-23,259,033) × 3 |
Likely Pathogenic |
| 27 |
37 |
6Y/M |
Obesity, Delayed development, Slow learner, EEG anomaly and ?Prader Willi Syndrome |
3 |
Gain |
20 |
q13.33 |
199 |
7 |
arr [hg19] 20q13.33 (62,624,900-62,824,265) × 3 |
Likely Pathogenic |
| 28 |
38 |
4Y/M |
Syndromic, Hyperactive, Impulsive, Speech delay |
1 |
Loss |
16 |
p12.2 |
775 |
3 |
arr [hg19] 22q11.22 (22,901,370-23,259,033) × 3 |
Pathogenic |
| 29 |
39 |
3 Years/F |
Developmental delay, Hypotonia, Dysmorphic features and Micrognathia |
3 |
Gain |
4 |
q35.2 |
435 |
2 |
arr [hg19] 4q35.2 (190,522,320-90,957,460) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
372 |
1 |
arr [hg19] 22q11.22 (22,929,364-23,301,460) × 3 |
Pathogenic |
| 30 |
41 |
45 Months/F |
Delayed Milestones |
3 |
Gain |
22 |
q11.22 |
341 |
1 |
arr [hg19] 22q11.22 (22,927,618-23,268,533) × 3 |
Pathogenic |
| 31 |
43 |
3Years/M |
Autism Spectrum Disorder |
3 |
Gain |
1 |
q11.22 |
22 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 32 |
44 |
3 Months/M |
Dysmorphic features, PDA syndrome and Radial Ray Anomaly |
3 |
Gain |
22 |
q11.22 |
358 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 33 |
45 |
9 Months/F |
Dysmorphic facies, Small mouth, Rocker bottom feet |
3 |
Gain |
3 |
q22.33 |
611 |
3 |
arr [hg19] 3q22.3q23 (138,215,113-38,825,892) × 3 |
Likely Pathogenic |
| |
|
|
|
1 |
Loss |
10 |
q11.22 |
1336 |
3 |
arr [hg19] 10q11.22 (46,989,206-48,203,688) × 3 |
Likely Pathogenic |
| |
|
|
|
4 |
Gain |
18 |
p11.32 |
18399 |
47 |
arr [hg19] 18p11.32q11.1 (136,227-18,534,784) × 4 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
358 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Likely Pathogenic |
| |
|
|
|
3 |
Gain |
X |
q28 |
75 |
4 |
arr [hg19] Xq28 (152,927,530-153,002,877) × 3 |
Likely Pathogenic |
| 34 |
46 |
3 Years/ M |
Autism |
3 |
Gain |
22 |
q11.22 |
358 |
2 |
arr [hg19] 22q11.22 (22,927,618-23,258,939) × 3 |
Pathogenic |
| 35 |
49 |
50 days/M |
Perinatal Hypoxia and Hypertonia |
3 |
Gain |
22 |
q11.22 |
331 |
1 |
arr [hg19] 22q11.22 (22,927,618-23,258,939) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q13.33 |
36 |
1 |
arr [hg19] 22q13.33 (51,092,246-51,127,901) × 3 |
Pathogenic |
| 36 |
57 |
15 years/m |
Convulsion and poor mental performance |
3 |
Gain |
22 |
q11.22 |
400 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,301,460) × 3 |
Pathogenic |
| 37 |
58 |
02Y/M |
Delayed Milestones |
3 |
Gain |
22 |
q11.22 |
377 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,278,835) × 3 |
Pathogenic |
| 38 |
59 |
02Y/M |
Autism, Speech delay and Hyperactivity |
3 |
Gain |
22 |
q11.22 |
400 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,301,460) × 3 |
Pathogenic |
| 39 |
60 |
03Y/M |
Macrocephaly and mild cortical artophy |
3 |
Gain |
22 |
q11.22 |
377 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,278,835) × 3 |
Pathogenic |
| 40 |
61 |
9 m/M |
Macrocephaly and mild cortical artophy |
3 |
Gain |
10 |
q11.22 |
1874 |
3 |
arr [hg19] 10q11.22 (46,293,590-48,167,553) × 4 |
Pathogenic |
| |
|
|
|
3 |
Gain |
17 |
p13.3 |
236 |
3 |
arr [hg19] 17p13.3 (811,480-1,047,339) × 3 |
Pathogenic |
| 41 |
62 |
5Y/m |
Autism Spectrum Disorder and Hyperactive |
1 |
Loss |
1 |
q21.1 |
121 |
2 |
arr [hg19] 1q21.1 (144,953,098-145,074,541) × 1 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
330 |
1 |
arr [hg19] 22q11.22 (22,929,364-23,258,939) × 3 |
Pathogenic |
| 42 |
68 |
5Y/m |
? Speech delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 43 |
69 |
7 Y/m |
Learning disability, Unable to understand simple things |
3 |
Gain |
22 |
q11.22 |
330 |
1 |
arr [hg19] 22q11.22 (22,929,364-23,258,939) × 3 |
Pathogenic |
| 44 |
72 |
32m/M |
Behavioural problem |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 45 |
74 |
4 Y/m |
Walking difficulty, developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 46 |
76 |
7 m/m |
Developmental delay, Hypotonia, Dysmorphic features |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 47 |
77 |
6 Y/M |
Global developmental delay, Dysmorphic features |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal male |
| 48 |
79 |
2 Y/F |
Global developmental delay with Epileptic encephalopathy and Dysmorphism |
1 |
Loss |
X |
p22.13 |
596 |
5 |
arr [hg19] Xp22.13(18,346,842-18,943,282) × 1 |
Pathogenic |
| 49 |
88 |
10Y/M |
? Facioscapulohumeral dystrophy, ? Myotonic muscular dystrophy |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 50 |
95 |
27 m/M |
Speech delay, Gross motor delay, Learning disability, Autism |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 51 |
98 |
6Y/M |
sm spectrum disorder, Developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 52 |
99 |
11 m/F |
Failure to thrive, IUGR |
1 |
Loss |
1 |
q21.1 |
906 |
8 |
arr [hg19] 1q21.1q21.2 (146,555,708-147,462,093) × 1 |
Pathogenic |
| |
|
|
|
1 |
Loss |
X |
p22.33 |
155065 |
711 |
arr [hg19] Xp22.33q28 (168,551-155,233,098) × 1 |
Pathogenic |
| 53 |
100 |
15 m/M |
Facial dysmorphism, Broad nasal bridge, B/L Microtia, Right facial palsy |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 54 |
104 |
4Y/M |
Developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 55 |
105 |
4Y/M |
MRI brain shows pansinusitis and adenoid hypertrophy |
3 |
Gain |
10 |
q11.12 |
2019 |
5 |
arr [hg19] 10q11.22 (46,252,072-48,270,746) × 3 |
Likely pathogenic |
| 56 |
108 |
9y/M |
History of consanguineous marriage of parents, febrile seizures, ? visual problem, loss of bowel |
3 |
Gain |
22 |
q11.22 |
400 |
3 |
arr [hg19] 22q11.22 (22,901,370-23,301,460) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
22 |
q11.22 |
400 |
3 |
arr [hg19] 22q11.22 (22,901,370-23,301,460) × 3 |
Pathogenic |
| 57 |
147 |
16 months/M |
Global developmental delay and Hypotonia |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 58 |
143 |
1y/M |
Dysmorphic features, low-level mosaic supernumerary marker chromosome detected in karyotype |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 59 |
154 |
2y/M |
ADHD with facial dysmorphism |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal male |
| 60 |
136 |
11Y/F |
Periodic fever |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 61 |
139 |
3y/M |
Autism spectrum disorder |
1 |
Loss |
15 |
q15.3 |
109 |
4 |
arr [hg19] 15q15.3 (43,868,570-43,977,181) × 1 |
Likely pathogenic |
| 62 |
141 |
7Y/F |
Developmental delay, short stature |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 63 |
137 |
2 months/F |
Severe pneumonia with dysmorphic facies |
3 |
Gain |
13 |
q14.11 |
73647 |
173 |
arr [hg19] 13q14.11q34 (41,460,947-115,107,733) × 3 |
Pathogenic |
| 64 |
129 |
32Y/F |
Autism spectrum disorder |
3 |
Gain |
15 |
q13.3 |
444 |
1 |
Arr [hg19] 15q13.3 (31,999,631-32,444,043) × 3 |
Pathogenic |
| |
|
|
|
3 |
Gain |
17 |
p13.3 |
211 |
3 |
arr [hg19] 17p13.3 (716,837-927,465) × 3 |
Pathogenic |
| 65 |
117 |
19 months/M |
Developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 66 |
115 |
3y/M |
Developmental delay, GDD with paternally inherited unbalanced t(7;10) |
1 |
Loss |
7 |
p22.33 |
2570 |
23 |
arr [hg19] 7p22.3 (43,376-2,613,293) × 1 |
Pathogenic |
| |
|
|
|
3 |
Gain |
10 |
q25.3 |
17841 |
79 |
arr [hg19] 10q25.3q26.3(117,585,175-135,426,386) × 3 |
Pathogenic |
| 67 |
138 |
4y/M |
Developmental delay, autistic features, seizures, dysmorphic course features |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 68 |
116 |
11y/M |
Assessment of language and hearing skill |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 69 |
119 |
3y/F |
Delayed development milestone |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 70 |
118 |
10 m/M |
Global developmental delay, Failure to thrive, Atrial Septal Defect |
1 |
Loss |
4 |
p16.3 |
4589 |
47 |
arr [hg19] 4p16.3p16.2 (68,345-4,656,856) × 1 |
Pathogenic |
| |
|
|
|
1.48 |
Loss mosaic |
4 |
p16.3 |
6368 |
55 |
arr [hg19] 4p16.3p16.1 (68,345-6,435,945) × 1-2 |
Pathogenic |
| 71 |
122 |
8 m/M |
Microcephaly, Strabismus, Failure to thrive |
1 |
Loss |
7 |
q11.23 |
1596 |
27 |
arr [hg19] 7q11.23 (72,621,345-74,217,791) × 1 |
Pathogenic |
| 72 |
125 |
22y/M |
Intellectual disability |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 73 |
155 |
14y/F |
Developmental delay, difficulty in walking, mild cerebellar atrophy |
2.2 |
Gain mosaic |
X |
p22.31 |
14203 |
67 |
arr [hg19] Xp22.31p22.12 (6,056,862-20,259,724) × 2-3 |
Pathogenic |
| 74 |
140 |
15y/M |
Seizures, Epilepsy |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 75 |
159 |
3y/M |
To rule out any chromosome abnormalities |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 76 |
152 |
2y/M |
Autism spectrum disorder |
|
|
|
|
|
|
arr (1-22) × 2, (XY) ×1 |
Normal male |
| 77 |
173 |
7y/F |
Developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 78 |
175 |
2y/M |
Microcephaly and Spasticity |
1.52 |
Loss mosaic |
22 |
q11.1 |
16417 |
165 |
arr [hg19] 22q11.1q12.3(16,888,899-33,305,441) × 1-2 |
Pathogenic |
| |
|
|
|
1 |
Loss |
22 |
q21.1 |
3152 |
44 |
arr [hg19] 22q11.21 (18,648,855-21,800,471) × 1 |
Pathogenic |
| 79 |
176 |
6y/M |
Facial Dysmorphism |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 80 |
177 |
4M/M |
?Leri weill dyschondrosteosis |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 81 |
179 |
2y/M |
Autism |
3 |
Gain |
22 |
q11.22 |
468 |
3 |
arr [hg19] 22q11.22 (22,833,258-23,301,460) × 3 |
Pathogenic |
| 82 |
180 |
15y/M |
Low IQ and Abnormal Facies |
2 |
Gain |
X |
q11.2 |
7072 |
39 |
arr [hg19] Xq11.2q13.1(63,706,926-70,779,158) × 2 |
Pathogenic |
| 83 |
181 |
14y/M |
Low IQ and Abnormal Facies |
2 |
Gain |
X |
q11.2 |
7072 |
39 |
arr [hg19] Xq11.2q13.1 (63,706,926-70,779,158) × 2 |
Pathogenic |
| 84 |
182 |
8y/M |
Intellectual Disability |
2 |
Gain |
X |
q11.2 |
7072 |
39 |
arr [hg19] Xq11.2q13.1 (63,706,926-70,779,158) ×2 |
Pathogenic |
| 85 |
186 |
11y/F |
Hearing disability and behaviour concern |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 86 |
188 |
3y/M |
Autism Spectrum Disorder |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal male |
| 87 |
197 |
08 Y/ m |
Learning disability and speech delay |
|
|
|
|
|
|
arr (1-22) ×2, (XY) ×1 |
Normal male |
| 88 |
198 |
20Y/F |
Sclerosis |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 89 |
199 |
10 Y/F |
Dysmorphism, Low set ears and poor coordination |
|
|
|
|
|
|
arr (1-22) ×2, (XX) ×1 |
High Long Continuous Regions of Homozygosity brother-half-sister parentage |
| 90 |
205 |
1Y/F |
Developmental delay, dysmorphic features and poor growth |
1.39 |
Loss mosaic |
14 |
q32.13 |
12,197 |
95 |
arr [hg19] 14q32.13q32.33 (95,087,352-107,284,437) × 1-2 |
Pathogenic |
| |
|
|
|
1 |
Loss |
14 |
q32.2 |
5,022 |
40 |
arr [hg19] 14q32.2q32.31 (97,163,311-102,185,159) × 1 |
Pathogenic |
| 91 |
209 |
2Y/M |
Dysmorphic facial features, hypotonia and failure to thrive |
3 |
Gain |
22 |
q11.22 |
358 |
2 |
arr [hg19] 22q11.22 (22,901,370-23,258,939) × 3 |
Pathogenic |
| 92 |
66 |
06Y/F |
Global developmental delay |
1.73 |
Loss Mosaic |
17 |
p13.2 |
29211 |
248 |
arr [hg19] 17p13.2q12 (5,239,141-34,450,123) × 1-2 |
Pathogenic |
| |
|
|
|
1 |
Loss |
17 |
p11.2 |
3688 |
35 |
arr [hg19] 17p11.2 (16,745,600-20,433,723) × 1 |
Pathogenic |
| 93 |
210 |
01Y/M |
Global developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 94 |
211 |
03Y/M |
Autism |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 95 |
212 |
05Y/M |
Intellectual Disability |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 96 |
213 |
07Y/M |
Delayed development milestone |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 97 |
214 |
03Y/M |
Autism, Speech delay and Hyperactivity |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 98 |
215 |
01Y/M |
Developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 99 |
216 |
07Y/M |
Delayed Milestones |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 100 |
217 |
09Y/M |
autism spectrum disorder |
|
|
|
|
|
|
arr (1-22) × 2, (XY) × 1 |
Normal Male |
| 101 |
218 |
02Y/F |
Intellectual Disability |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |
| 102 |
219 |
04Y/F |
Global developmental delay |
|
|
|
|
|
|
arr (1-22) × 2, (XX) × 1 |
Normal female |