Jing-Xia Li1,2, Sakura Hosoba1,3, Harris AC Wayne1, Amanda C Box4, James R Spivey5, Kyle T Bradley6 and Edmund K Waller1*
1Department of Hematology and Medical Oncology, Winship Cancer Institute, Emory University, Atlanta, GA, USA
2Department of Respiratory Medicine, Xiangya Hospital, Central South University, Changsha, Hunan, China
3Department of Internal Medicine, Division of Hematology, Shiga University of Medical Science, Otsu, Japan
4Department of Hematology and Medical Oncology-BMT, Emory University, Atlanta, GA, USA
5Department of Medicine and the Emory Transplant Center, Emory University Hospital, Atlanta, GA, USA
6Department of Pathology and Laboratory Medicine, Emory University, Atlanta, GA, USA
- Corresponding Author:
- Edmund K Waller
1365B Clifton Rd. RoomB5305, Atlanta, GA 30322, USA
Tel: 0014047274995
E-mail: ewaller@emory.edu
Received date: March 15, 2016; Accepted date: April 18, 2016; Published date: April 22, 2016
Citation: Li JX, Hosoba S, Wayne HAC, et al. Classical Hodgkin Lymphoma Presenting as A Post-Transplant Lymphoproliferative Disorder after Organ Allograft Transplantation. Transl Biomed. 2016, 7:2.
Keywords
Hodgkin lymphoma; Post-transplant lymphoproliferative disorder; Organ allograft transplantation
Introduction
Post-transplant lymphoproliferative disorder (PTLD) is a serious complication arising from immunosuppression after solid organ transplantation (SOT) or hematopoietic stem cell transplantation (HSCT). The incidence of PTLD ranges from 1%-8% within SOT recipients [1,2]. A peak incidence at less than 6 months post-transplant is usually seen after allogeneic HSCT (allo-HSCT), while SOT PTLD usually presents several months to years after organ transplant [3]. PTLDs after SOT are mostly recipient-derived, while PTLDs after allo-HSCT arise from donor lymphoid cells [3]. Among the various histologic types of PTLD, classical Hodgkin lymphoma PTLD (cHL-PTLD) represents only 1.8-3.4% of all PTLD cases [2,4,5], but is reported to have more favorable outcome than cases of cHLlike PTLD in which the large Reed Sternberg-like cells have an immunophenotype that deviates from typical cHL, including expression of CD45 and CD20, and absence of CD15 [5,6].
In this report, we review the published experience in the management of 22 cases of cHL-PTLD after SOT in adults and based upon the principles of care from the successful treatment of an institutional case of cHL-PTLD after liver transplantation.
Methods
Literature and institutional database review: previous case reports and case series of adult cHL-PTLD were collected from PubMed from 1974-2015 using the search terms “Hodgkin lymphoma/disease”, “post-transplant lymphoproliferative disorder/disease” and “solid organ transplant”, and the results are summarized in Table 1. Data on recipients were extracted from each report including age, sex, organ graft, time between transplant and diagnosis, immunosuppression, Epstein-Barr Virus (EBV) status, treatment regimen, response and survival, and follow-up. Under an IRB-approved protocol, analysis of our institutional database of SOT performed between 2004 and 2012 at Emory showed that among 2911 cases of transplants, the single patient described in the current report is the sole case of cHL-PTLD. This case of cHL-PTLD and all of the cases from the literature were pathologically confirmed. The survival curve (Figure 1) was generated using the Kaplan-Meier method.
Age/
Sex |
Organ graft |
Time+ (mo) |
Immunosuppression |
EBV |
Treatment regimen |
Response/Survival |
Follow-up (Mo) |
Year |
| 48/M |
Kidney |
29 |
AZA, Pred |
ND |
RI, MOPP |
NR/dead |
3 |
1974 |
| 23/F |
Kidney |
68 |
AZA, Pred |
ND |
MOPP, bleomycin, Surgery |
ND/alive |
22 |
1977 |
| 27/M |
Kidney |
85 |
CSA, AZA, Pred |
ND |
ND |
ND/dead |
0.5 |
1977 |
| 55/M |
Kidney |
42 |
AZA, mPSL |
ND |
RI, MOPP, then ABVD |
ND/dead |
4 |
1983 |
| 26/F |
Liver |
55 |
CSA, Pred |
+ |
RI, MOPP |
CR/alive |
15 |
1993 |
| 42/M |
Kidney |
84 |
CSA, AZA, Pred |
+ |
RI, ABV, RT |
CR/alive |
19 |
1994 |
| 44/M |
Kidney |
51 |
CSA, AZA, Pred |
- |
RI, ChlVPP |
CR/alive |
61 |
1996 |
| 41/M |
Kidney |
26 |
CSA, AZA, Pred |
+ |
RI, RT |
CR/alive |
18 |
1996 |
| 26/M |
Heart |
60 |
CSA, AZA |
+ |
RI, ChlVPP, RT |
CR/alive |
12 |
1996 |
| 24/M |
Kidney |
19 |
CSA, AZA, Steroid |
+ |
RI, MOPP-ABVD, RT |
Relapse/alive |
25 |
1996 |
| 30/M |
Kidney |
20 |
CSA, AZA, Steroid |
+ |
RI, ABV, Surgery |
CR/dead |
31 |
1996 |
| 24/M |
Kidney |
28 |
CSA, AZA, Steroid |
+ |
RI, ABVD, RT, Surgery |
CR/alive |
67 |
1996 |
| 34/M |
Kidney |
87 |
CSA, AZA, Steroid |
+ |
RI, ABVD, RT, Surgery |
CR/dead |
7 |
1996 |
| 42/M |
Kidney |
81 |
CSA, AZA, Steroid |
+ |
RI, ABV, RT, Surgery |
CR/alive |
18 |
1996 |
| 32/M |
Kidney |
51 |
CSA, AZA, Steroid |
+ |
RI |
ND/ND |
ND |
1996 |
| 31/M |
Heart/lung |
78 |
CSA |
+ |
ChlVPP |
Poor/dead |
14 |
1996 |
| 54/M |
Heart |
108 |
CSA |
+ |
ChlVPP |
PR/alive |
12 |
1996 |
| 24/M |
Kidney |
108 |
CSA, MMF |
+ |
RI, R, ABVD |
CR/alive |
6 |
2005 |
| 41/M |
Kidney |
120 |
CSA, MMF, Pred |
+ |
RI, ABVD, RT |
CR/alive |
8 |
2006 |
| 27/M |
Kidney |
84 |
CSA, AZA, Pred |
+ |
RI, ABVD |
Relapse/alive |
108 |
2006 |
| 46/M |
Kidney |
122 |
CSA, AZA, Pred |
+ |
RI, Chemo, RT |
CR/alive |
94 |
2015 |
| 51/M |
Kidney |
90 |
CSA, MMF, Pred |
ND |
RI, Chemo |
CR/alive |
22 |
2015 |
| 66/F |
Liver |
4 |
TAC, MMF, Pred |
+ |
RI, R-ChlVPP-AV |
CR/alive |
9 |
2015, current report |
Abbreviations: ABVD, Adriamycin, Bleomycin, Vinblastine, and Dacarbazine; ABV, Adriamycin, Bleomycin, and Vinblastine; AV, Adriamycin and Vincrinstine; Allo-HSCT, Allogeneic Hematopoietic Stem Cell Transplantation; AZA, Azathioprine; Chemo, Chemotherapy; cHL, Classical Hodgkin Lymphoma; ChlVPP, Chlorambucil, Vinblastine, Procarbazine, and Prednisone; CMV: Cytomegalovirus; CNI, Calcineurin Inhibitor; CR, Complete Remission; CSA, Cyclosporine A; EBER, Epstein-Barr Virus-encoded RNA; EBNA, Epstein-Barr Virus Nuclear Antigen; EBV, Epstein-Barr Virus; F, Female; FDG-PET, 18F-Fluorodeoxyglucose -Positron Emission Tomography; LMP, Latent Membrane Protein; M, Male; MMF, Mycophenolate Mofetil; Mo, Month; MOPP, Nitrogen Mustard, Vincristine, Procarbazine, Prednisone; mPSL, Methylprednisolone; ND, Not Documented; NHL, non-Hodgkin lymphoma; NR, No Response; PR, Partial Remission; Pred, Prednisone; PTLD, Post- Transplant Lymphoproliferative Disorder; R, Rituximab; RI, Reduction of Immunosuppression; RT, Radiotherapy; SOT, Solid Organ Transplantation; TAC, Tacrolimus
Table 1: cHL-PTLD after solid organ transplant reported in literature
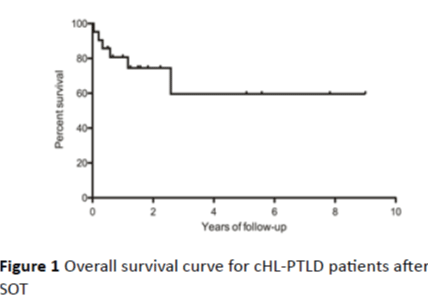
Figure 1: Overall survival curve for cHL-PTLD patients after SOT
Results
Patient characteristics
Among the 22 cases reported in the literature and the single case from our institution, 87% were male and 13% were female, with a median age of 34 years (range: 23-66 years). Patients with cHL-PTLD had received allografts of kidney (78%), heart/lung (13%) and liver (9%). The median interval between transplant and diagnosis of cHL-PTLD was 68 months (range: 4-122 months), which is consistent with late onset of PTLD in SOT. Evidence for EBV positivity at the level of EBV RNA or protein was found in 74% of the cases. All of the patients with cHL-PTLD were receiving immunosuppressant at the time of diagnosis: 65% were receiving a triple-drug immunosuppressive regimen consisting of a calcineurin inhibitor (CNI), an anti-metabolite and steroids. Cyclosporine A (CSA, 83%) was the most commonly used CNI. Azathioprine (AZA, 70%) or mycophenolate mofetil (MMF, 17%) were the commonly used anti-metabolites. Steroids were administered in 83% of the patients, usually in addition to CNI and/or an anti-metabolite. Among the cases that have defined cHL subtypes, mixed cellularity cHL (56.25%) was most common, followed by nodular sclerosis (31.25%) and lymphocyte predominant (12.5%) subtypes. The treatment of cHL-PTLD in published reports varied. Reduction of immunosuppression (RI) occurred in 83% of the patients as initial therapy. Other treatments described in the literature include rituximab, chemotherapy, immune therapy and local therapy such as radiation and surgery [2,5,7]. In this series, almost half of the patients received radiotherapy, and one fifth of them had surgical resection. MOPP (Nitrogen Mustard, vincristine, procarbazine, prednisone) and ChlVPP (chlorambucil, vinblastine, procarbazine, and prednisone) were used prior to 1996, but these regimens were then replaced or supplemented by ABV/ABVD (Adriamycin, bleomycin, vinblastine, with or without dacarbazine). However, the clinical outcome seen in patients treated with different regimens was equivalent, with an overall survival of 64% at 9 years (Figure 1).
Institutional Experience
With this background, we developed a treatment plan for a 66-year-old female with a diagnosis of cHL-PTLD that developed after a liver transplant from a deceased donor for treatment of cryptogenic cirrhosis. Both the recipient and the donor had positive EBV serology before transplant. Posttransplant immunosuppressive drug therapy consisted of prednisone, MMF and Tacrolimus (TAC). At four months posttransplant, she developed swelling and pain in the right parotid gland, on stable doses of MMF and TAC with no history of organ graft rejection. Parotidectomy revealed EBV-positive cHL-PTLD with typical morphologic and immunophenotypic features including numerous large mononuclear Hodgkin cells and binucleated Reed-Sternberg cells in a T-cell–rich background (Figure 2A). Eosinophils were sparse. Immunohistochemical stains demonstrated positive staining of the Hodgkin and Reed-Sternberg cells for CD30, CD15, PAX5 (dim), and CD20 (minor subset) and negative staining for CD45, CD3, EMA and ALK1 (Figures 2B-2D). The large cells were positive for EBV-encoded RNA (EBER) by in situ hybridization (Figure 2E). Two of two peri-parotid lymph nodes were also involved. An 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET) scan showed foci of FDG uptake in the chest, abdomen, pelvis and multiple bone sites. A bone marrow biopsy showed normal cellularity with no evidence of lymphoma.
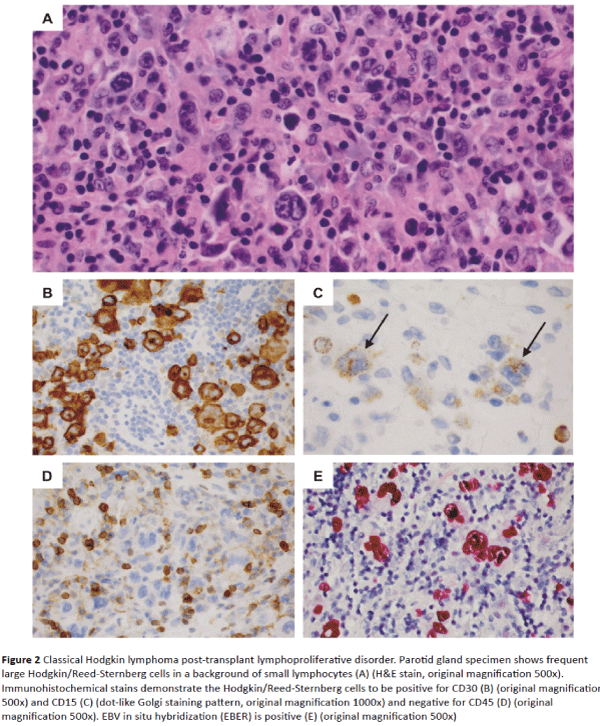
Figure 2: Classical Hodgkin lymphoma post-transplant lymphoproliferative disorder. Parotid gland specimen shows frequent large Hodgkin/Reed-Sternberg cells in a background of small lymphocytes (A) (H&E stain, original magnification 500x). Immunohistochemical stains demonstrate the Hodgkin/Reed-Sternberg cells to be positive for CD30 (B) (original magnification 500x) and CD15 (C) (dot-like Golgi staining pattern, original magnification 1000x) and negative for CD45 (D) (original magnification 500x). EBV in situ hybridization (EBER) is positive (E) (original magnification 500x)
Based on the literature, we treated this patient with a 28- day combination chemotherapy regimen consisting of rituximab 375 mg/m2 on day 1, chlorambucil 6 mg/m2/day on day 1 to day 7, vinblastine 6 mg/m2 on day 1, procarbazine 100 mg/m2/day on day 1 to day 7, prednisolone 40 mg/day on days 1 to 14, and doxorubicin 25 mg/m2 and vincristine 1.4 mg/m2 on day 8 (R-ChlVPP-AV) [8]. Bleomycin was not included in this regimen due to potential pulmonary toxicity. Rituximab was added to the chemotherapy regimen given expression of CD20 on a subset of malignant cells. In total, the patient completed 4 cycles of chemotherapy and achieved a clinical, radiographic and scintigraphic complete remission (CR), without any episodes of liver rejection or infections, that
Eight months after the last cycle of R-ChlVPP-AV combination therapy, EBV-DNA was undetectable and a restaging FDG-PET scan showed CR with no abnormal FDG uptake. Cytomegalovirus (CMV) viral load has been monitored closely and was undetected throughout the treatment. Immunosuppression during chemotherapy consisted of single agent TAC, as MMF and prednisone were tapered off given the relation between the level of immune suppression and PTLD. TAC was later replaced by cyclosporine due to concerns about TAC contributing to persistent neutropenia (Figure 3A). EBVDNA levels dropped from 1900 IU/ml to 880 IU/ml after starting treatment with rituximab and chemotherapy, and reducing the immunosuppression to single agent TAC. EBVDNA level was undetectable at 9 months follow-up after completing chemotherapy. Grade 3 regimen-related neutropenia (neutrophils 500-1,000/mm3) was noted which was responsive to granulocyte colony stimulating factor. A post-therapy bone marrow biopsy showed normal cellularity without evidence of myelodysplasia or lymphoma. Neutropenia has gradually resolved over the 9 months after completion of treatment.
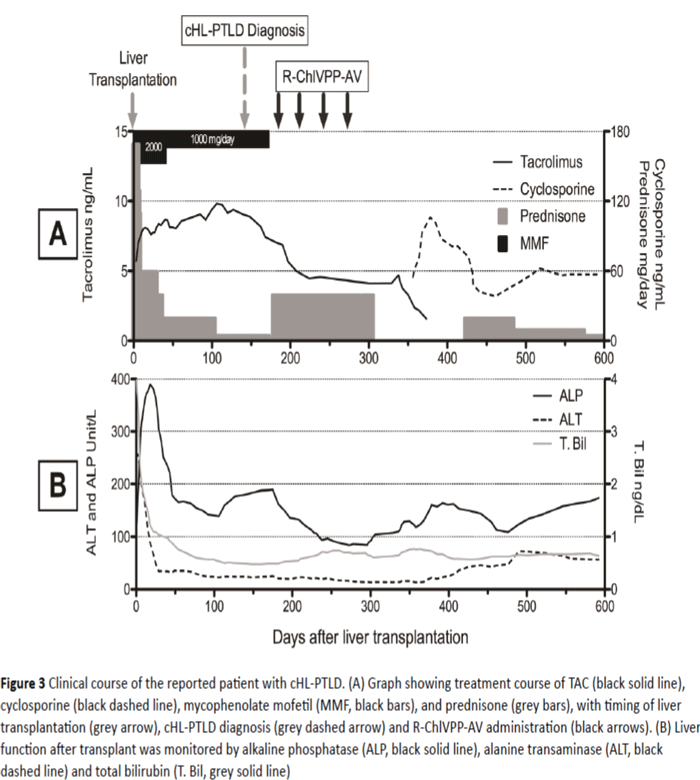
Figure 3: Clinical course of the reported patient with cHL-PTLD. (A) Graph showing treatment course of TAC (black solid line), cyclosporine (black dashed line), mycophenolate mofetil (MMF, black bars), and prednisone (grey bars), with timing of liver transplantation (grey arrow), cHL-PTLD diagnosis (grey dashed arrow) and R-ChlVPP-AV administration (black arrows). (B) Liver function after transplant was monitored by alkaline phosphatase (ALP, black solid line), alanine transaminase (ALT, black dashed line) and total bilirubin (T. Bil, grey solid line)
Discussion
The literature review of cHL-PTLD revealed relatively few reports of this disorder compared with more than 100 reports describing other types of PTLD, supporting the rarity of this disease entity, and consistent with a reported frequency of less than 4% of all PTLD cases [2,4,5]. There is no evidence of association between CMV and the development of PTLD as discussed by Kim and colleagues [9]. While both cHL and non- Hodgkin lymphoma (NHL) PTLD share EBV as a possible causal agent, the low frequency of EBV-cHL-PTLD suggests that normal immune responses directed to EBV-encoded antigens may be sufficient to control or eliminate this entity, even in the setting of post-transplant immunosuppression. Most individuals who have had prior EBV infection have relatively high frequencies of memory effector T cells that target EBVencoded peptides expressed on HLA class I or class II proteins. EBV can infect B cells and exhibits different patterns of EBV gene or protein expression that are classified as one of three latency programs [10]. Type 1 latency expresses EBV nuclear antigen (EBNA) 1 and EBER dominantly and is associated with Burkitt lymphoma. Type 2 latency is characterized by expression of EBNA1, latent membrane protein (LMP) 1 and LMP2. Normally, an EBV-infected atypical post-germinal center B cell that lacks a functional B-cell receptor would be expected to undergo apoptosis. However, due to expression of EBNA1, LMP1 and LMP2, an EBV-infected B-cell can survive negative selection and develop into the Hodgkin/Reed-Sternberg cells seen in cHL [11]. In type 3 latency, all the latent antigens (EBNA1, 2, 3A, 3B, 3C, and LP and LMP1, LMP2A and LMP2B) are expressed. This type of latency is associated with EBVrelated NHL PTLD.
Due to the role of EBV in the etiology of PTLD, anti-viral drugs are often given post-transplant to limit EBV reactivation and early events associated with the proliferation of EBVinfected cells. Prophylactic anti-viral use following transplantation reduces the risk of PTLD risk in renal transplant recipients by up to 83% [12]. The combination of monitoring EBV-DNA viral load and adjusting immunosuppression accordingly is associated with a lower incidence of PTLD following lung transplantation [13].
Of note, while the institutional patient described above achieved a CR with 4 cycles of outpatient treatment and was never hospitalized with febrile neutropenia, she did experience significant grade 3 neutropenia during treatment that persisted for 7 months after her last cycle of treatment. Recovery from neutropenia persisted after completion of chemotherapy in spite of normal levels of B1, B6, B12, Vitamin D, and copper in the blood, and the absence of anti-neutrophil antibodies. Neutrophil recovery was coincident with the expected clearance of rituximab, supporting a potential role for B-cells in myelopoiesis that has been noted in other cases of rituximab-related neutropenia [14]. Given the very good response of cHL-PTLD based upon cases reviewed from the literature, a less intensive regimen with less hematopoietic toxicity may have been equally effective.
In conclusion, cHL-PTLD is a rare subtype of PTLD, which often presents as late onset and is EBV-associated. Treatment of cHL-PTLD is compatible with preservation of graft function. Despite its rarity, cHL-PTLD responds well to involved field radiation and combination chemotherapy, and has a relatively favorable prognosis.
9540
References
- Ashrafi F, Shahidi S, Ebrahimi Z, Mortazavi M (2015) Outcome of rapamycin therapy for post-transplant-lymphoproliferative disorder after kidney transplantation: case series. Int J Hematol Oncol Stem Cell Res 9: 26-32.
- Choi JH, Ahn MJ, Oh YH, Han SW, Kim HJ, et al. (2006) Epstein-Barr virus-associated Hodgkin's disease following renal transplantation. Korean J Intern Med 21: 46-49.
- Novoa-Takara L, Perkins SL, Qi D, ShidhamVB, Vesole DH, et al. (2005) Histogenetic Phenotypes of B Cells in Posttransplant Lymphoproliferative Disorders by Immunohistochemical Analysis Correlate With Transplant Type. Am J Clin Pathol 123: 104-112.
- Pitman SD, Huang Q, Zuppan CW, Rowsell EH, Cao JD, et al. (2006) Hodgkin lymphoma-like posttransplant lymphoproliferative disorder (HL-like PTLD) simulates monomorphic B-cell PTLD both clinically and pathologically. Am J Surg Pathol 30: 470-476.
- Stern M, Herrmann R, Rochlitz C, Dirnhofer S, Pless M (2005) A case of postÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂtransplant lymphoproliferative disease presenting as CD20ÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂexpressing, Epstein–BarrÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂvirus positive Hodgkin lymphoma. Eur J Haematol 74: 267-270.
- Basso S, Zecca M, Calafiore L, Rubert L, Fiocchi R, et al. (2013) Successful treatment of a classic Hodgkin lymphomaÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂtype postÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂtransplant lymphoproliferative disorder with tailored chemotherapy and Epstein–Barr virusÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂspecific cytotoxic T lymphocytes in a pediatric heart transplant recipient. Pediatr Transplant 17: E168-E173.
- Flanagan KH, Brennan DC (2006) EBVÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂassociated recurrent Hodgkin's disease after renal transplantation. Transpl Int 19: 338-341.
- Evens AM, Sweettenham JW, Horning SJ (2008) Hodgkin lymphoma in older patients: an uncommon disease in need of study. Oncology 22: 1369-1369.
- Kim JM, Lee SK, Kim SJ, Joh JW, Kwon CHD, et al. (2010) Risk factors for posttransplant lymphoproliferative disorder in pediatric liver transplant recipients with cytomegalovirus antigenemia. Transplant Proc 42: 895-899.
- Grywalska E, Rolinski J (2015) Epstein-Barr Virus–Associated Lymphomas. Semin Oncol 42: 291-303.
- Vockerodt M, Cader FZ, Shannon-Lowe C, Murray P (2014) Epstein-Barr virus and the origin of Hodgkin lymphoma. Chin J Cancer 33: 591-597.
- Funch DP, Walker AM, Schneider G, Ziyadeh NJ, Pescovitz MD (2005) Ganciclovir and Acyclovir Reduce the Risk of PostÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂTransplant Lymphoproliferative Disorder in Renal Transplant Recipients. Am J Transplant 5: 2894-2900.
- Bakker NA, Verschuuren EA, Erasmus ME, Hepkema BG, Veeger NJ, et al. (2007). Epstein-Barr Virus–DNA Load Monitoring Late After Lung Transplantation: A Surrogate Marker of the Degree of Immunosuppression and a Safe Guide to Reduce Immunosuppression. Transplantation 83: 433-438.
- Dunleavy K, Tay K, Wilson WH (2010) Rituximab-associated neutropenia. Semin Hematol 47: 180-18

