Key words
Diabetes mellitus, cognition, memory, insulin, cortisol.
List of abbreviations
DM, diabetes mellitus; T2DM: type 2 diabetes mellitus; T1DM: type 1 diabetes mellitus; LHPA: limbic-hypothalamic-pituitary-adrenal axis; GC: glucocorticoids; CRH: corticotropin-releasing hormone; ACTH: adrencorticotropic hormone; HbAI: glycolysated hemoglobin; BMI: body mass index; HOMA-I: homeostatic model assessment to quantify insulin resistance; TC: total cholesterol; TG: triglycerides; LDL-c: low density lipoprotein cholesterol; HDL-c: high density lipoprotein cholesterol; ERPs: Event Related Potentials; DEX: dexamethasone; MMSE: Mini-Mental State Examination; SBST: Stanford Binet subtests testing; WMS-R: Wechsler Memory Scale-Revised; STZ: streptozotocin.
Introduction
Type 2 diabetes mellitus (T2DM) is characterized by abnormalities in carbohydrate and fat metabolism, chronic hyperglycemia, insulin resistance, and a relative insulin secretion defect. T2DM comprises 90% of DM [1]. Many organ systems are adversely affected by diabetes, including the brain, which undergoes changes that may increase the risk of cognitive decline (as in memory, attention, executive function, and psychomotor speed) [2,3]. It is likely that DM-induced cognitive decline has a multifactorial etiology through multiple mechanisms including blood glucose and direct effect of chronic hyperglycemia on the brain [4], blood lipid [5], blood pressure [6], insulin resistance [7], hypoglycemia [8], chronic complication as micro and macro-vascular complications (diabetic vasculopathy) [9], dysregulation of limbic-hypothalamic- pituitary-adrenal axis (LHPA) (stress response) [10, 11], advanced glycation end products, inflammatory cytokines, oxidative stress [12] and diabetes-related depression [13,14]. It has been suggested that insulin antagonistic action of hormones and neuropeptides like catecholamines (adrenaline, noradrenaline and dopamine), GCs, sex steroids and adipokines as well as dysregulation of autonomic nervous activity may contribute to the early development of insulin resistance (pre-diabetic condition) and T2DM [15]. Some of these factors can directly impair glucose uptake capacity and this might be due to alterations in key proteins involved in insulin’s intracellular signaling pathways [16]. Glucocorticoids (GCs) and glucose regulation are closely linked. In T2DM, several recent studies point to the dysregulation of the LHPA with disturbances in glucose regulation leading to hypercortisolemia [10,11]. Under physiological conditions, corticotropin-releasing hormone (CRH) from the hypothalamus leads to adrencorticotropic hormone (ACTH) release from the pituitary. This in turn stimulates secretion of GCs from the adrenal cortex. By acting on a wide array of target tissues, GCs are important for successful adaptation (stress response). Once elevated, GCs exert a negative feedback via the pituitary, hypothalamus and hippocampus [17]. GCs modulate a broad range of neural functions, including stress reactivity and emotional responses; neuronal excitability and plasticity, learning and memory, neurogenesis and neuronal death. Normal brain function requires optimal levels of the GCs signaling and that deviation from this optimal level in either direction is highly deleterious [18, 19]. Although acute cortisol elevations during stress are protective, chronically elevated levels have mostly negative effects [17]. Disturbance in glucose regulation as insulin resistance and T2DM are associated with hypercortisolemia [10-20]. Chronic hyperglycemia and hypercortisolemia are known to have deleterious effects on the hippocampus [17], an area of the brain responsible for learning and memory (particularly episodic and declarative memory and especially important for accurate and reliable contextual memory) [21] and also with the highest colocalization of in insulin [22], glucocorticoid [21-23] and glutamate [24] receptors. However, reports on the relationship between hypercortisolemia and cognitive impairment in patients with hyperglycemia are few or even inconsistent and controversial.
Aim of the work
In this study, we aimed to determine the relationship between hypercortisolemia and cognitive function in patients with T2DM. We therefore undertook an integral approach consisted of the followings: 1) evaluation of cognitive function of patients with T2DM using a battery of cognitive testing which are able to determine mild impairment in different cognitive domains, 2) We determined confounders associated with cognitive function with T2DM including: age, duration of illness, hypertension, dyslipidemia, body mass index (BMI), insulin resistance, level of glycemic control (as indicative by glycolysated hemoglobin (HbAIc) and cortisol levels at basal conditions and with dexamethasone (DEX) suppression test, and 3) Linear regression analysis was done to determine the relationship between cortisol levels and scores of cognitive testing after controlling of related confounders.
Materials and Methods
This is a cross-sectional study included 57 patients with T2DM. Patients were randomly recruited from the Internal Medicine and Neurology departments of Assiut and Al-Alzar University Hospitals. The diagnosis of DM was made according to the World Health Organization Expert Committee on DM, Geneva: WHO [25]. Diabetics met one or more of the following criteria: 1) had a fasting glucose value greater than 125 mg/dl on two separate occasions; 2) had a 2 hours glucose value greater than 200 mg/dl during a 75 gram oral glucose tolerance test; or 3) had a prior diagnosis of T2DM, and were being treated with hypoglycemic agents and/or diet and exercise. Forty-ages-, sex-, socioeconomic status- and educationally- matched subjects were included in this study as healthy controls for comparison. Control subjects were recruited from the general population. The protocol of this study was in conformity with the local ethical guidelines and informed written consent was obtained from each participant. All study participants underwent a standardized interview questionnaire regarding vascular risk factors. Excluded from this study were subjects with: 1) known medical illness other than DM and its associated dyslipidemia, hypertension, insulin resistance and obesity) 2) history of hypoglycemic coma or complications of DM (other than peripheral neuropathy) as nephropathy, retinopathy, etc; 3) primary neurological condition as history of transient ischemic attacks, cerebrovascular stroke or epilepsy or psychiatric disease (other than depression); 4) previous serious head injury; 5) any sensory or motor disorder that would preclude psychological testing (including blindness); 6) regular treatment with any medications other than insulin and/or hypoglycemic drugs, or medication known to have psychoactive effects such as benzodiazepines, beta-adrenoceptor antagonists, steroids, major tranquillizers and antidepressants; 7) drug or alcohol abuse; and 8) smoking.
Data collection
Demographic and clinical data were collected as follow: age, gender, systolic blood pressure, diastolic blood pressure, weight, height, BMI. Weight was measured to the nearest 0.1 kg on a calibrated balance beam scale. Height was measured to the nearest 0.5 cm by a tape measure. BMI was calculated as follow: BMI [weight (kg)/height (m2)]. Normal was defined if BMI is >20 to <25 kg/m2, over weight if BMI is equal or >25 to <30, obese if BMI is equal or >30 to <35 and morbidly obese if BMI is >35 [26]. Hypertension was defined according to the National Cholesterol Education Program guidelines [27]. The following criteria were used: 1) systolic value ≥130 mm Hg, 2) diastolic value ≥85 mm Hg, or 3) use of antihypertensive medication. Dyslipidemia was also defined following National Cholesterol Education Program guidelines. The following criteria were used: 1) statin treatment, 2) triglyceride levels ≥150 mg/dl, or 3) high-density lipoprotein levels ≤40 mg/dl for men and ≤50 mg/dl for women.
Specimen collection and analysis
Venous blood samples were drawn from patients at 8.00 a.m. Routine hematology tests were done and included: fasting blood glucose (FBG), complete blood count (CBC), renal function, lipogram (serum total cholesterol (TC), triglycerides (TG), low density lipoprotein cholesterol (LDL-c), high density lipoprotein cholesterol (HDL-c)) and uric acid. Serum levels of TC, TG, HDL-c and LDL-c were measured by enzymatic colourimetric method using the autoanalyzer Hitachi 911 (Boehinger, Mannheim, USA). Serum uric acid was determined by colorimetric US plus kit, supplied by Roche diagnostics, (GmbH, D-68298 Mannheim, USA). Plasma levels of glucose, insulin, and HbA1c were assessed after an overnight fast. The patients received a standard lunch (light balanced diet: 600 kcal and formed of 35% protein, 30% fat and 35% carbohydrates). Two hours after meal, 3 ml blood samples were withdrawn from all participants for estimated of post-prandial serum glucose (PBG) level. Insulin was determined in duplicate using an enzyme-linked immunosorbent assay (ELISA) (Diagnostic Systems Laboratory, Webster, TX. USA). Insulin resistance was calculated using the homeostasis model assessment equation for insulin resistance (HOMA-IR) formula as follow: Fasting insulin (uU/mL) X fasting glucose (mmol/L) /22.5. Patients were consider to have insulin resistance if HOMA-IR ≥ 2.6 [28]. For estimation of the basal cortisol level, two independent blood samples were collected within 2 min of each other just before the glucose ingestion during a standardized glucose tolerance test. They were averaged and used as an estimate of basal cortisol secretion. Total cortisol was measured with an enzyme immunoassay (EIA; IBL, Hamburg, Germany). In DEX suppression test, 1 mg of dexamethasone was given to the patient at 11 p.m., and blood was withdrawn at 8 a.m. for a cortisol measurement [29]. DEX is a human-made (synthetic) steroid that is similar to cortisol. Therefore, taking dexamethasone should reduce ACTH levels in normal people and lead to decrease in cortisol levels while increasing cortisol levels after dexamethasone indicates abnormal response.
Cognitive assessment
Cognitive functions were assessed independently for each participant by two experienced psychologists and under supervision of the psychiatrist, using a set of standardized Arabic translated neuropsychological tests which are sensitive for mild cognitive impairment and covering different cognitive domains. They included: Mini-Mental State Examination (MMSE) [29,30], Stanford Binet 10 subsets testing (SBST) (4th edition) [31,32] and Wechsler Memory Scale-Revised (WMSR) [33]. From SBST, we selected vocabulary and comprehension for assessment of verbal reasoning, pattern analysis for assessment of visual reasoning, quantitation for quantitative reasoning, and bead memory and memory for sentences for short-term memory. From WMS-R, we tested digit forward, digit backward, mental control, associate learning, logical memory and visual reproduction.
Event-Related Potentials (ERPs) testing
Before examining ERPs, all participants underwent basic audiological testing (Amplaid Model 720, Milan, Italy). Testing for ERPs was done on a separate day after completion of neuropsychological testing (Neuropack S1 EMG/EP measuring system, MEB-9400 (Nihon Kohden, Japan). ERPs are series of scalp waves that are extracted from the electroencephalogram (EEG) by time domain analysis and averaging of EEG activity following multiple stimulus repetitions. They were elicited with an auditory discrimination task paradigm by presenting a series of biaural 1000 Hz (standard) versus 2000 Hz (target) tones at 70 dB with a 10 millisecond rise/fall and 40 millisecond plateau time. P300, the late component of ERPs was obtained. Latencies and amplitudes (peak to peak) of P300 component of ERPs were measured. P300 is believed to index stimulus significance and the amount of attention allocated to the eliciting stimulus event, being maximal to task-relevant or attended stimuli and being absent or small to task-irrelevant or unattended stimuli [35].
Psychological evaluation
Standardized psychiatric interview was done by applying the Diagnostic and Statistical Manual of Mental Health Disorders, fourth edition (DSM–IV) criteria for the diagnosis of depression [36]. A differentiation between clinical depression and depressive symptoms was made throughout this work. The Arabic version [37] of the Beck Depression Inventory (BDI-II) [38] was used for assessment of the severity of depressive symptoms. BDI–II items are in alignment with DSM–IV criteria. BDI–II consists of 21 items, each corresponds to a symptom of depression is summed to give a single score for the BDI-II. According to that scale, the patient may have, no or minimal depressive symptoms (scoring: 0-13), mild symptoms (scoring: 14-19), moderate symptoms (scoring: 20-28), and severe symptoms (scoring: 29-63).
Statistical analysis
Calculations were done with the statistical package of SPSS, version 12.0. Data were presented as mean±SD (standard deviation) as they were normally distributed. Unpaired twosided Student’s t test was used for comparison of means. Correlations between score of cognitive testing and diabetes related demographic, clinical, other risk factors, level of glycemic control; depression scores and hormones were assessed using Pearson’s test. To determine the relationship between cortisol levels and cognitive function in patients with T2DM, linear regressions analysis was done using total score of cognition testing as the dependent variable. Age was controlled as covariants (entered as the first step). Cortisol levels after DEX was entered as the second step. HbA1c (Indicative of level of glycemic control) was added as the third step to ascertain whether long-term glycemic control would add to the variance explained by the cortisol after DEX and we then inverted steps two and three to determine the amount of variance explained by cortisol level after DEX after taking HbA1c into account. For all tests, values of p<0.05 were considered statistically significant.
Results
This study included 57 patients with T2DM (male = 20; females = 37), with mean age of 39.57±13.89 years and duration of illness of 7.37±5.15 years. Nearly half of the patients were uncontrolled on anti-diabetic treatment. Demographic and clinical characteristics of the studied group were shown in tables (1). Table (2) showed comparison between patients and control subjects in scores of cognitive functions and BDI-II for depression. Patients had significantly lower scores of MMSE, different subsets of SBST, WMS-R and total scores of cognitive testing (MMSE, SBST and WMS-R) (P=0.004) and higher scores of BDI-II (P=0.001) regardless to the degree of control on anti-diabetic medications. In this study, nearly 75.43% (n = 43) of patients with T2DM had depressive symptomatology, mostly of mild/moderate type (52.63%; n = 30). Table (3) showed comparison between patients and control subjects in ERPs variables in relation to the degree of control on anti-diabetic medications. Patients had significantly prolonged latencies (P=0.0001) and reduced amplitudes (P=0.0001) of P300 component of ERPs regardless to the degree of control on anti-diabetic medications (with the exception of P300 amplitude on the right side). Table (4) showed comparison between patients and controls in concentrations of glucose, HbIAc, insulin and cortisol at basal conditions and after DEX in relation to the degree of control on anti-diabetic medications. Patient had significantly higher concentrations of fasting and post-prandial glucose level, HbA1c, insulin and basal cortisol and cortisol after DEX,
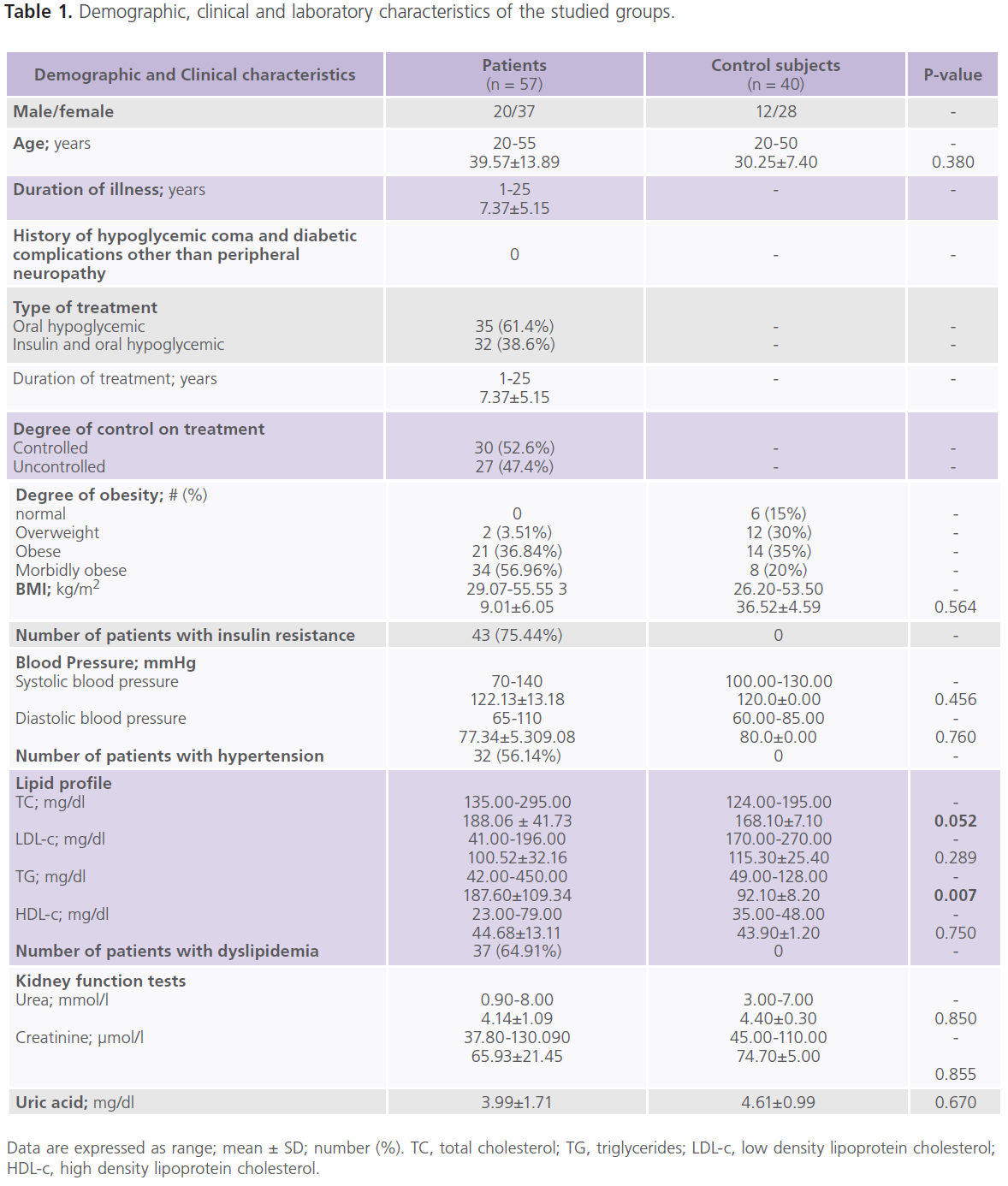
Table 1: Demographic, clinical and laboratory characteristics of the studied groups.
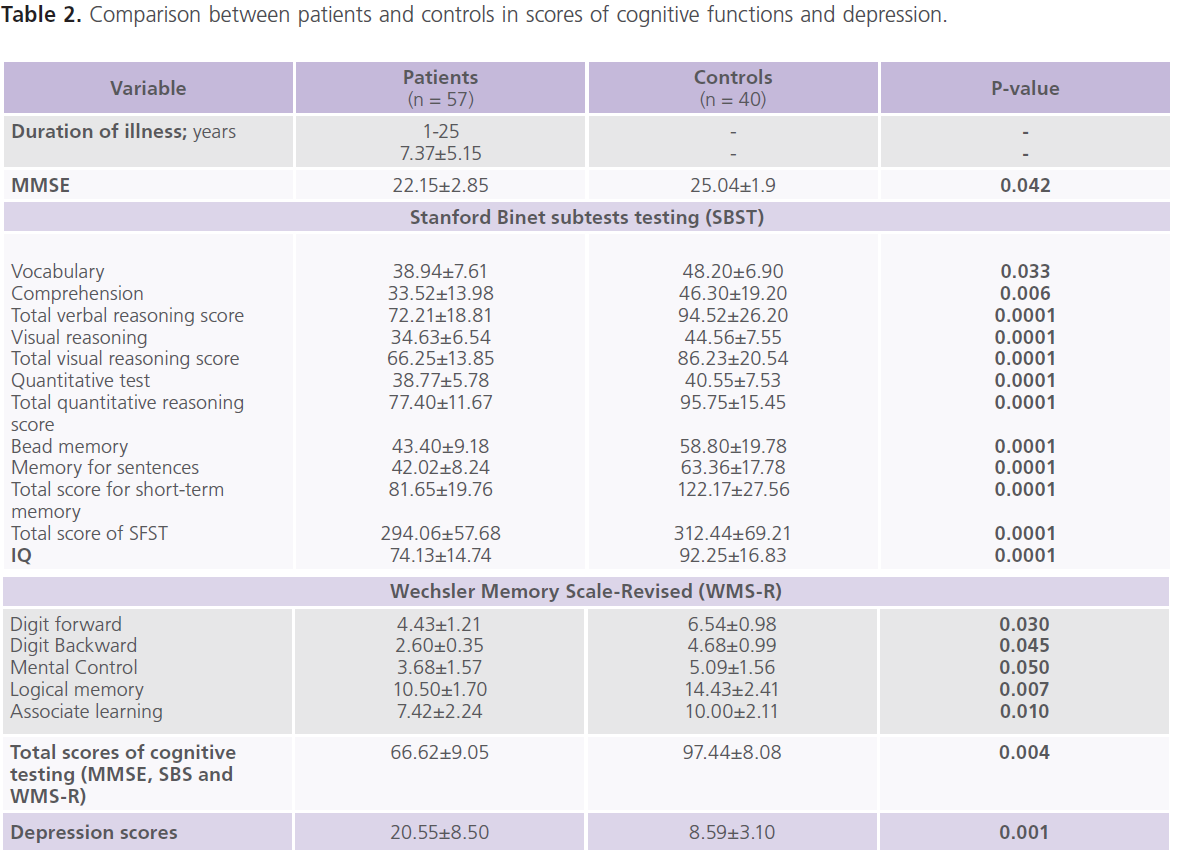
Table 2: Comparison between patients and controls in scores of cognitive functions and depression.
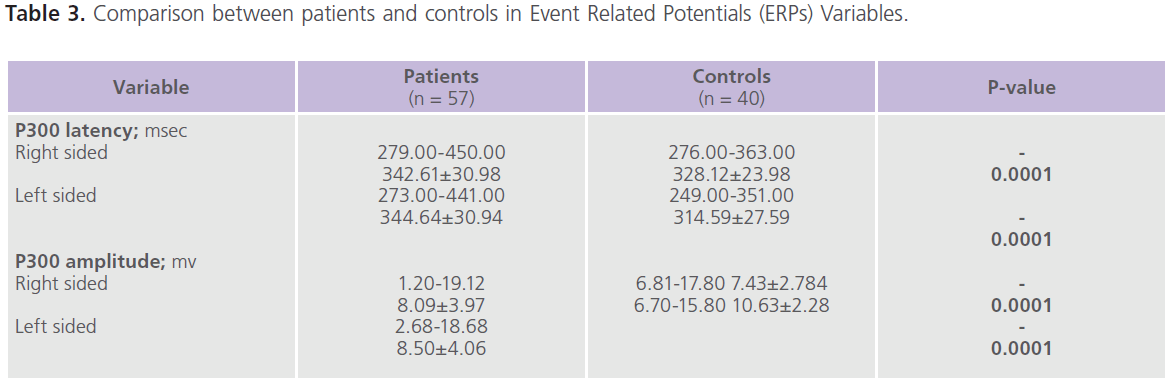
Table 3: Comparison between patients and controls in Event Related Potentials (ERPs) Variables.
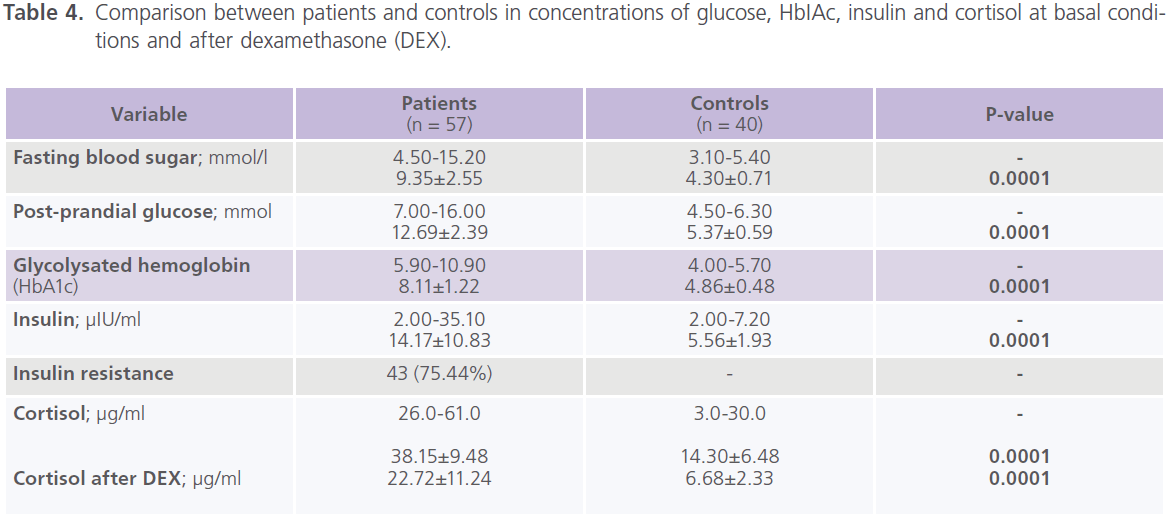
Table 4: Comparison between patients and controls in concentrations of glucose, HbIAc, insulin and cortisol at basal conditions and after dexamethasone (DEX).
particularly those who were uncontrolled on anti-diabetic medications. Table (5) showed significant correlations between total scores of cognitive testing and age (r = -0.370, p = 0.050), duration of illness (r = -0.658, p = 0.001), blood glucose level (r = -0.543, p = 0.010) and ERPs latency (r = -0.560, p = 0.004) and amplitude (r = 0.340, p = 0.053), insulin (r = -0.365, p = 0.004) and cortisol (r = -0.456, p = 0.001). BMI was positively correlated with HbA1c (r = 0.372, p = 0.021); HbA1c was positively correlated with cortisol (r = 0.341, p = 0.036) and HOMA-IR was positively correlated with insulin (r = 0.914, p = 0.001) and cortisol (r = 0.364, p = 0.025) concentrations. Table (6) showed the associations between cognition, cortisol levels after DEX test, and HbA1c derived from linear regression analyses. We observed an association between total scores of cognitive testing (MMSE, SBST and WMR-S) and higher cortisol levels after DEX only in association with poor glycemic control but disappeared with glycemic control.
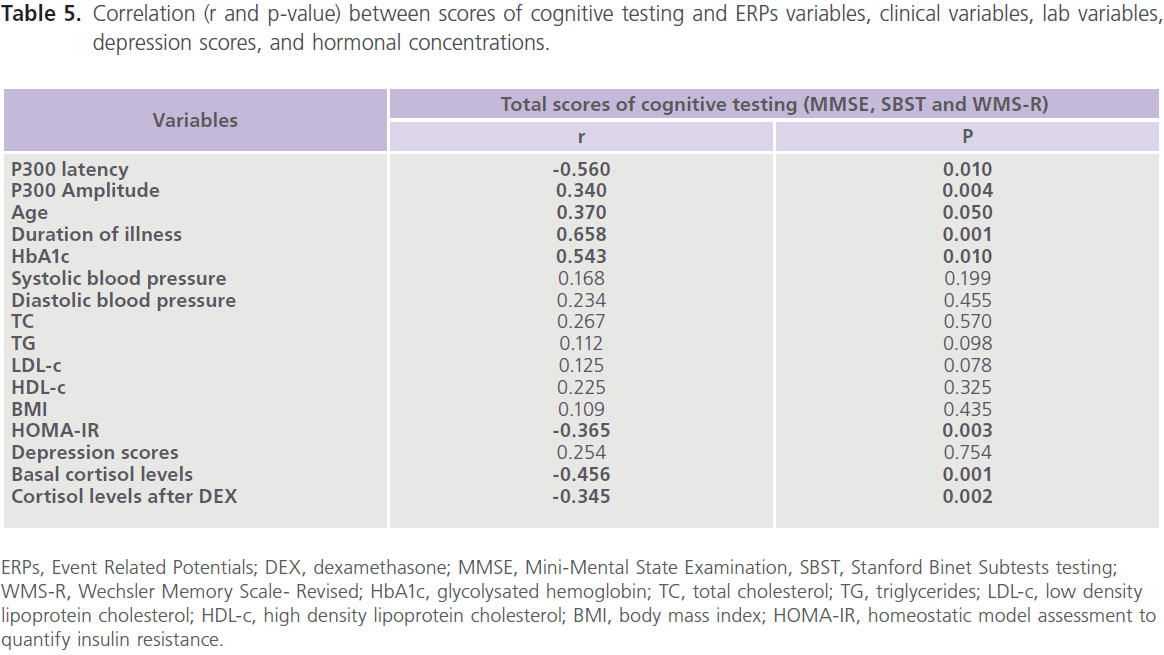
Table 5: Correlation (r and p-value) between scores of cognitive testing and ERPs variables, clinical variables, lab variables, depression scores, and hormonal concentrations.
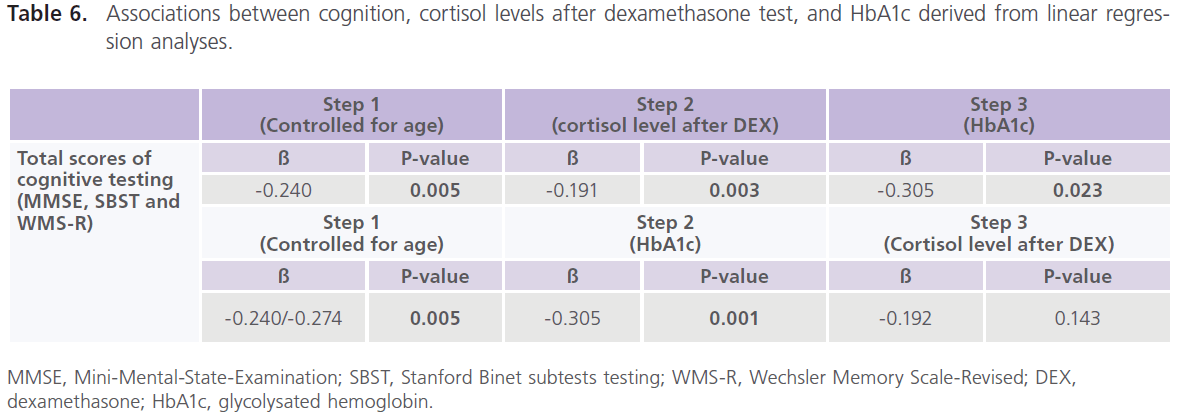
Table 6: Associations between cognition, cortisol levels after dexamethasone test, and HbA1c derived from linear regression analyses.
Discussion
Evidences from this study further confirm the risk of cognitive dysfunction with T2DM as follow: Patients with T2DM had low scores of cognitive testing and poor performance in different cognitive tasks as verbal relations, comprehension, visual reasoning, pattern analysis, quantitation, bead memory, short term memory and memory for sentences, digit forward, digit backward, mental control, logical memory and associate learning and abnormalities in P300 component of ERPs, a physiological analogue of cognitive testing [30]. Total scores of cognitive testing was found to be negatively associated with, age. The presence of insulin resistance, the duration of DM and overall level of glycemic control as indicated by HbAIc [40, 41]. In accordance, Cox et al. [40] observed that the increase of blood glucose >15mmol/l was associated with marked decline in cognition and poor performance in arithmetic tasks. Wu et al. [41]. observed that compared to treated patients, the untreated patients with DM had 2 points decline over 2 years on the MMSE with a duration of illness <5 years and a 6 point decrease on the MMSE with a duration of illness ≥ 5 years. In addition, many authors also observed an improvement in performance of cognitive tests with improvement in glucose tolerance [40, 41]. Hippocampal abnormalities, an area of the brain responsible for learning and memory, were incriminated as a cause of cognitive dysfunction observed among patients with chronic DM. Studies demonstrated that well-controlled middle-aged individuals with T2DM and non-diabetic individuals with insulin resistance have declarative memory deficits and specific hippocampal volume reductions. It has been suggested that the decreases in hippocampal insulin receptors activities in T2DM contributed to behavioral deficits in type 2 rodents and cognitive deficits in humans.
In the present study, a high frequency of patients had insulin resistance (75.44%), hypertension (56%) and dyslipidemia (65%), however, none were related to poor cognitive scores. Depression was also frequently reported (75%), but was not related to poor cognitive scores. Epidemiological studies suggested that diabetic patients are two to three folds more likely to develop depressive illness when compared to nondiabetic individuals. In general, the prevalence of depression with DM was estimated to be 31.1% [42].
In the present study, T2DM was associated with hypercortisolemia at the basal conditions and after DEX administrations (DEX suppression test) particularly in patients with poor glycemic control. In accordance, several studies done on patients with disturbed glucose regulation as with insulin resistance and T2DM, observed elevation of cortisol levels at basal measures [10], after DEX suppression test [43] and after CRH administration [44]. Others observed association between cortisol level and glycemic control [10]. While in contrast, others reported no alterations [45]. The variable results across studies may be attributed to different methodologies and populations studied. In the study, the relationship between hypercortisolemia after DEX remained significant only in association with poor glycemic control but disappeared with glycemic control. In accordance, Bruehl et al. [11] observed an association between impairment in declarative memory, elevated basal plasma cortisol levels, higher levels after DEX suppression, a larger response to CRH, positive association between HbAIc and cortisol levels during the DEX/CRH test independent of age, BMI, hypertension, and dyslipidemia. the authors also observed an inverse association between declarative memory and cortisol levels across all subjects and with glycemic control. From the results of this study and others and the pathophysiological similarities between DM and stress-related cognitive deficits and mood disorders, we suggest that T2DM is a common metabolic disorder accompanied by cognitive deficits, depression and increased secretion of adrenal stress hormone cortisol (hypercorticism), in which all are influenced by the level of glycemic control. We suggest that the anatomically related structures as the hippocampus which is implicated in cognitive deficits and the amygdala which is implicated in the activation of stress response, are very sensitive to glycemic control and might be responsible for the cognitive deficits and depression associated with T2DM [13,14,46]. This is further supported by the following observations: First: the specific role of the hippocampus, in cognitive dysfunction, stress, mood disorders and aging have been investigated in a number of animal models and human studies in T1DM [23], T2DM [11], stress related neuropsychiatric disorders and age related cognitive deficits and behavioral abnormalities [13,14]. Stranahan et al. [46] showed that rats treated with streptozotocin (STZ, animal model of T1DM) had hypoinsulinemia, hyperglycemia, hypercortisolemia, and impairments in hippocampal neurogenesis, synaptic plasticity and learning. Similar deficits are observed in db/db mice, which are characterized by insulin resistance, elevated corticosterone and obesity. Revsin et al. [23] observed obvious cognitive deficits in the novel object-placement recognition task streptozotocin (STZ) mice. The authors observed that hippocampal alterations in STZ-diabetic mice and chronic excess of glucocorticoid secretion and was characterized by: a) increased glial fibrillary acidic protein-positive astrocytes as a marker reacting to neurodegeneration, b) increased c-Jun expression marking neuronal activation and reduced Ki-67 immunostaining indicating decreased cell proliferation, and c) blockade of glucoreceptors GR resulted in attenuation of the morphological signs of hippocampal aberrations and rescued the diabetic mice from the cognitive deficits. Second: The LHPA is also activated in people with T2DM and there are data linking increased cortisol concentrations with cognitive impairment observed of type 1 and type 2 diabetic animals [23]. On the other hand, GCs have been implicated as pathophysiological mediators of obesity, insulin resistance and T2DM [11,23, 47]. Gene expression profiling revealed a significant down-regulation of several glutamate receptor subunits and glutamate transporters in the hippocampus with functional damage to the hippocampal cells of GRov mice, a very distinctive animal model with overexpression of GRs specifically in forebrain and had characteristic phenotype with increased reactivity to the environment along with emotional lability, increase in anxiety-like and depression-like behaviors was shown [48]. Third: Glutamate signaling is also essential in controlling the structural and functional plasticity of the synapse and plays a critical role in learning and memory mechanisms within the hippocampus [49]. A large body of work demonstrates the key role of altered glutamatergic mechanisms in glucocorticoid-induced cognitive deficits and deficits associated with normal aging. Several age-related changes in glutamatergic function have been described, including a reduced capacity for glutamate uptake and a loss in the number of high-affinity transporters in the terminals of aged animals and age associated decreases in the hippocampal density of glutamatergic receptors and cognitive consequences [24]. Aging is also associated with dysregulation of LHPA, leading to a delayed termination of the stress response and in turn increases exposure to GCs which exacerbates the likelihood of neural damage, particularly in the hippocampus. Furthermore, previous studies have observed an exaggerated LHPA response to this challenge test among depressed patients as well as among the elderly due to reduction in feedback regulation due to a hippocampal GC receptor deficit [17]. Forth: Insulin is a competitive inhibitor for insulin degrading enzyme [50]. and thus persistent elevations in insulin,as may interfere with peripheral AB amyloid, clearance and this could lead to higher AB concentrations in the brain [51]. Another possibility is that chronically high insulin in the periphery may paradoxically causes a relative hypoinsulinized state in the brain and hyperinsulinemia could actually result in cognitive dysfunction by impairing insulin-mediated utilization of glucose by cells in the brain particularly the hippocampus, which is enriched with insulin receptors [22]. The knowledge that cognitive deficits are frequently associated with T2DM will have important implications for treatment of T2DM and for research purposes. Future researches have to include the following: a) longitudinal studies that prospectively assess the relation of the disease process to cognition over time, b) comprehensive longitudinal evaluation of endocrine status, cognition, and brain imaging, and 3) randomized clinical trials that compare cognitive function in DM patients receiving memory enhancers, antidepressants, versus a control group of DM patients. However, and despite the strength of our findings, this study had some limitations which include: 1) a relatively small sample size; 2) we did not measure ACTH to determine the link between T2DM and LHPA regulation.
Conclusions
In patients with T2DM, hypercortisolemia appears to exacerbate cognition dysfunction only in presence of poor glycemic control. Large-scale epidemiological and intervention studies might enhance our understanding and management of diabetes-related cognitive and behavioral abnormalities.
Competing interests
We declare that this work has no conflict of interests. There is no involvement of sponsor for this work design, data collection, analysis, interpretation, drafting, nor the decision to submit this paper for publication. All are authors’ responsibility.
Acknowledgement and funding
We thank the paramedical staff members of the out-patient clinic of the diabetes mellitus unit of the Internal Medicine department of Assiut University Hospital, Assiut, Egypt, for their help throughout the study. Authors were responsible for the work design, data collection, analysis, interpretation, drafting, the decision to submit this paper for publication.
6466
References
- Bonow, RO., Gheorghiade, M. The diabetes epidemic: a national and global crisis. Am J Med. 2004; 116 (Suppl. 5A): 2S-10S.
- Ryan, CM., Geckl, MO. Circumscribed cognitive dysfunction in middleaged adults with type 2 diabetes. Diabetes Care 2000; 23 (10): 1486- 1493.
- Cosway, R., Strachan, MW., Dougall, A., Frier, BM., Deary, IJ. Cognitive function and information processing in type 2 diabetes. Diabet Mellitus 2001; 18 (10): 803-810.
- Awad, N., Gagnon, M., Messier, C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J Clin Exp Neuropsychol 2004; 26 (8): 1044-1080.
- Henderson, VW., Guthrie, JR., Dennerstein, L. Serum lipids and memory in a population based cohort of middle age women. J Neurol Neurosurg Psychiatry 2003; 74 (11): 1530-1535.
- Hassing, LB., Hofer, SM., Nilsson, SE., Berg, S., Pedersen, NL., McClearn, G., Johansson, B. Comorbid type 2 diabetes mellitus and hypertension exacerbates cognitive decline: evidence from a longitudinal study. Age Ageing 2004; 33 (4): 355-361.
- Taylor, VH., MacQueen, GM. Cognitive dysfunction associated with metabolic syndrome. Obes Rev. 2007; 8 (5): 409-418.
- Draelos, MT., Jacobson, AM., Weinger, K., Widom, B., Ryan, CM., Finkelstein, DM., Simonson, DC. Cognitive function in patients with insulin-dependent diabetes mellitus during hyperglycemia and hypoglycemia. Am J Med. 1995; 98 (2): 135-144.
- Ryan, CM., Geckle, MO., Orchard, TJ. Cognitive efficiency declines over time in adults with Type 1 diabetes: effects of micro- and macrovascular complications. Diabetologia 2003; 46 (7): 940-948.
- Lee, ZS., Chan, JC., Yeung, VT., Chow, CC., Lau, MS., Ko, GT., Li, JK., Cockram, CS., Critchley, JA. Plasma insulin, growth hormone, cortisol, and central obesity among young Chinese type 2 diabetic patients. Diabetes Care 1999; 22 (9): 1450-1457.
- Bruehl, H., Rueger, M., Dziobek, I., Sweat, V., Tirsi, A., Javier, E., Arentoft, A., Wolf, OT., Convit, A. Hypothalamic-pituitary-adrenal axis dysregulation and memory impairments in type 2 diabetes. J Clin Endocrinol Metab. 2007; 92 (7): 2439-2445.
- Rösen, P., Nawroth, PP., King, G., Möller, W., Tritschler, HJ., Packer, L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCOMCBN, the American Diabetes Association and the German Diabetes Society. Diabetes/Metab Res Rev. 2001; 17 (3): 189-212.
- McEwen, BS., Magarinos, AM., Reagan, LP. Studies of hormone action in the hippocampal formation: possible relevance to depression and diabetes. J Psychosom Res. 2002; 53 (4): 883-890.
- Lustman, PJ., Clouse, RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications 2005; 19 (2): 113-122.
- Burén, J., Eriksson, JW. Is insulin resistance caused by defects in insulin’s target cells or by a stressed mind? Diabetes Metab Res Rev. 2005; 21 (6): 487-494.
- Dagogo-Jack, S., Liu, J., Askari, H., Tykodi, G., Umamaheswaran, I. Impaired leptin response to glucocorticoid as a chronic complication of diabetes. J Diabetes Complications 2000; 14 (6): 327-332.
- Heuser, I., Lammers, CH. Stress and the brain. Neurobiol Aging 2003; 24 (supl. 1): S69-S76.
- Diamond, DM., Bennett, MC., Fleshner, M., Rose, GM. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 1992; 2 (4): 421-430.
- de Kloet, ER., Joëls, M., Holsboer, F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005; 6 (6): 463-475.
- Rosmond, R. Stress induced disturbances of the HPA axis: a pathway to Type 2 diabetes? Med Sci Monit. 2003; 9 (2): RA35-RA39.
- Jacobson, L., Sapolsky, RM. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev. 1991; 12 (2): 118-134.
- Zhao, WQ., Alkon, DL. Role of insulin and insulin receptor in learning and memory. Molecular and Cellular Endocrinology 2001; 177 (1-2): 125-134.
- Revsin, Y., Rekers, NV., Louwe, MC., Saravia, FE., De Nicola, AF., de Kloet, ER., Oitzl, MS. Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocininduced type 1 diabetes mice. Neuropsychopharmacology 2009; 34 (3): 747-758.
- Segovia, G., Porras, A., Del Arco, A., Mora, F. Glutamatergic neurotransmission in aging: a critical perspective. Mech Ageing Dev. 2001; 122 (1): 1-29.
- WHO. Expert Committee on Biological Standardization. Thirty-fifth report. World Health Organ Tech Rep Ser. 1985; 725: 1-140.
- Niewoehner CB. Endocrine pathophysiology. 1998. Fence Ceek Publ., Madison.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection. Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Circulation 2002; 106 (25): 3143-3421.
- Matthews, DR., Hosker, JP., Rudenski, AS., Naylor, BA., Treacher, DF., Turner, RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28 (7): 41–-419.
- Stewart, PM. The adrenal cortex: Kronenberg, HM., Melmed, S., Polonsky, KS., Larsen, P . eds. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders Elsevier. 2008.
- Folstein, MF., Folstein, SE., McHugh, PH. “Mini-mental state” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975; 12 (3): 189-198.
- Al-Rajeh, S., Ogunniyi, A., Awada, A., Daif, A., Zaidan, R. Preliminary assessment of an Arabic version of the mini-mental state examination. Annals of Saudi Medicine 1999; 19 (2): 150-152.
- Delany, EA., Hopkins, TF. The Stanford. Binet Intelligence scale: fourth Edition: Examiner’s Handbook. Chicago. The Riverside Publishing Co. 1986.
- Melika, LK. The Stanford Binet Intelligence Scale, fourth edition. Arabic Examiner’s Handbook. Cairo: Dar El Maref Publishing; Egypt, Cairo. 1998.
- Wechsler, D. Wechsler Memory Scales-Revised. New York: Psychological cooperation. 1987.
- Polich, J. P300 clinical utility and control of variability. J Clin Neurophysio.1998; 15 (1): 14-33.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (4th ed.) Washington, DC: American Psychiatric Association. 1994: 317-391.
- Gharyb, AG. Beck Depression Inventory–II (BDI-II), Arabic Examiner’s Handbook. Dar El -Anglo Publishing, Egypt, Cairo. 2000.
- Beck, AT., Steer, RA., Ball, R., Ranieri, W. Comparison of Beck Depression Inventories -IA and -II in psychiatric outpatient”. Journal of personality assessment 1996; 67 (3): 588-597.
- Kurita, A., Mochio, S., Isogai, Y. Changes in auditory P300 event related potentials and brainstem evoked potentials in diabetes mellitus. Acta Neurol Scand. 1995; 92 (4): 319-323.
- Cox, DJ., Kovatchev, BP., Gonder-Frederick, LA., Summers, KH., McCall A, Grimm KJ, Clarke WL. Relationships between hyperglycemia and cognitive performance among adults with type 1 and type 2 diabetes. Diabetes Care 2005; 28 (1): 71-77.
- Wu, JH., Haan, MN., Liang, J., Ghosh, D., Gonzalez, HM., Herman, WH. Impact of diabetes on cognitive function among older Latinos: a population-based cohort study. J Clin Epidemiol. 2003; 56 (7): 686- 693.
- Shehatah, A., Rabie, MA., Al-Shahry, A. Prevalence and correlates of depressive disorders in elderly with type 2 diabetes in primary health care settings. J Affect Disord. 2101; 123 (1-3): 197-201.
- Catargi, B., Rigalleau, V., Poussin, A., Ronci-Chaix, N., Bex, V., Vergnot, V., Gin, H., Roger, P., Tabarin, A. Occult Cushing’s syndrome in type-2 diabetes. J Clin Endocrinal Metab. 2003; 88 (12): 5808-5813.
- Fernández-Real, JM., Engel, WR., Simó, R., Salinas, I., Webb, SM. Study of glucose tolerance in consecutive patients harbouring incidental adrenal tumors. Study Group of Incidental Adrenal Adenoma. Clin Endocrinol (Oxf) 1998; 49 (1): 53-61..
- Andrews, RC., Herlihy, O., Livingstone, DEW., Andrew, R., Walker, BR. Abnormal cortisol metabolism and tissue sensitivity to cortisol in patients with glucose intolerance. J Clin Endocrinal Metab. 2002; 87 (12): 5587-5593.
- Stranahan, AM., Arumugam, TV., Cutler, RG., Lee, K., Egan, JM., Mattson, MP. Diabetes impairs hippocampal function through glucocorticoid mediated effects on new and mature neurons. Nat Neurosci. 2008; 11 (3): 309-317.
- den Heijer, T., Vermeer, SE., van Dijk, EJ., Prins, ND., Koudstaal, PJ., Hofman, A., Breteler, MM. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia; 46 (12): 1604- 1610.
- Wei, Q., Lu, XY., Liu, L., Schafer, G., Shieh, KR., Burke, S., Robinson, TE., Watson, SJ., Seasholtz, AF., Akilm, H. Glucocorticoid receptor overexpression in forebrain: a mouse model of increased emotional lability. Proc Natl Acad Sci USA 2004; 101 (32): 11851-11856.
- Kim, JJ., Diamond, DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 2002; 3 (6): 453-462.
- Duckworth, WC., Bennett, RG., Hamel, FG. Insulin degradation: Progress and potential. Endocr Rev. 1998; 19 (15): 608-624.
- Fishel, MA., Watson, GS., Montine, TJ., Wang, Q., Green, PS., Kulstad, JJ., Cook, DG., Peskind, ER., Baker, LD., Goldgaber, D., Nie, W..et al. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch Neurol. 2005; 62 (10): 1539-1544.

