Keywords
Myasthenia gravis, cognition, autonomic neuropathy; nicotinic acetyl choline receptors; thymoma
Abbreviations
MG, myasthenia gravis; NMJ, neuromuscular junction; nAChRs, nicotinic acetylcholine receptors; ACh, acetyl choline; Abs, antibodies; AChE-Is, acetylcholinesterase inhibitors; CSF, cerebrospinal fluid; EAMG, experimental autoimmune myasthenia gravis; EPP, end plate potential; IVIGs, intravenous immunoglobulins; SM, skeletal muscles; anti-SM Abs, anti-striated muscles antibodies; EEG, electroencephalography; CSA, central sleep apnea; OSA, obstructive sleep apnea; AAN, acute autonomic neuropathy; NE, norepinephrine; EEG, electroencephalography; MIR, main immunogenic region; CNS, central nervous system; LEMS, Lambert-Eaton myasthenic gravis; Th cells, T helper cells; ANNA-1, antineuronal antibody nuclear type 1; SCLC, small cell lung cancer; EAAN, experimental acute autonomic neuropathy; APCs, antigen presenting cells
Introduction
Myasthenia gravis (MG) is an immune-mediated neuromuscular junction (NMJ) disorder characterized by muscle fatigue and weakness peaking at the end of the day. MG is mainly caused by antibodies (Abs) against muscle nicotinic acetylcholine receptors (nAChRs) at the postsynaptic membrane resulting in depletion of acetylcholine (ACh) at the NMJ (1). While the prevailing clinical finding of MG is muscle fatigue and weakness, rarely patients may develop additional nervous system manifestations and syndromes as memory difficulties (2-4), sleep abnormalities (5-7) and autonomic dysfunction (8-10). Furthermore, certain nervous system disorders may coexist in patients with MG as epilepsy (11,12), peripheral neuropathy (10,13), multiple sclerosis (14-17), dermatomyositis (18), neuromyelitis optica (19,20), dementia (21) and psychiatric disorders (22-24). Several central and peripheral mechanisms have to be suggested for the association between MG and nervous system manifestations and disorders. Involvement of cholinergic nervous systems and pathways by well-known and others not well known immune mediated processes resulting in generalized cholinergic deficiency is a highly suggested mechanism (4,7,10,16,25). This is based on the following observations: 1) The structural identities between different muscle and neuronal nAChRs subunits with the possibility of cross-reactivity between different nAChRs Abs (26), and 2) some cancers (e.g. thymoma and small cell lung cancer) express immune responses driven by muscle and neuronal nAChR subtypes which account for several related paraneoplastic neurological disorders affecting the cholinergic systems (27,28). However, controversial views claims against the view of generalized cholinergic deficiency in MG and suggest that the comorbid nervous system manifestations with MG may result from: 1) a response against non-specifically acting cytokines or as a non-specific multiple autoimmune response in presence or absence of tumor (29), or 2) consequences (or complications) of MG (as mood disorder, respiratory impairment, hypoxia, sleep abnormalities and adverse effects from acetylcholine esterase inhibitors (AChE-Is) (30-32).
The present article serves as an overview of recent studies in MG literature present in pubmed which highlighted comorbid or associated nervous and non-nervous system diseases (publications till 2011 were checked). We also checked the reference lists of retrieved studies for additional reports, in addition to our experience in this field. In this article, we reviewed manifestations of central, peripheral and autonomic nervous system involvement or syndromes present in patients with MG. Then we discussed the possible mechanisms that may underlie such co-morbidities based on: the structure of different nAChRs subtypes in MG and its comorbid immunerelated diseases of the nervous system, immunopathogenesis of MG, tumor immunology and non-specific autoimmunity, and the complications of MG and its medications. We believe that recognition of co-morbidities with MG is mandatory, not only for diagnosis, determining prognosis and management but also for future advances in understanding the cellular and molecular mechanisms of MG and its immunopathogenic spectrum for proper therapeutic strategies.
Basic Information
Myasthenia gravis (MG) is an uncommon disease with annual incidence varies from 2-5/106 to 10.4/106, and its prevalence vary from 25/106 to 125/106. The disease tends to affect women more often than men (3:2). Women are usually affected between the ages of 20 and 40, but after the age of 50, both sexes tend to be equally affected (33-35). The diagnosis of MG is made according to the clinical, pharamacologic, electrophysiologic and immunologic criteria. Fatigability is the cardinal symptom. Fatigue increases following physical exertion (i.e., exhaustion and tiredness) and resolves after a period of rest. Muscle fatigue and weakness initially involve the ocular muscles in about 2/3 of patients then spread to the bulbar and limb muscles (36). Approximately 85% of patients develop generalized weakness while symptoms remain confined to the extraocular muscles in about 10% of patients. Many patients progress from mild to severe disease. Table 1 showed the two commonly and practically used classification systems for MG. Osserman’s clinical classification divides patients according to the affected muscle groups and the intensity of involvement (37). In 1997, the Medical Scientific Advisory Board (MSAB) of the MG Foundation of America (MGFA) clinically classifies and grade patients for the use in therapeutic research trials (38). In severe forms of MG and its complications as myasthenic crisis with and without acute oropharyngeal dysfunction, the weakness of respiratory muscles may become severe enough to require mechanical ventilation. Approximately 12-16% of patients with MG will experience a crisis episode (39). Spontaneous remissions are very rare and last for varying periods which mostly occur during the first 3 years of the disease (40).

Table 1: Clinical classification of myasthenia gravis

Figure 1: The Medical Scientific Advisory Board (MSAB) of the MG Foundation of America (MGFA)
Diagnostically, muscle weakness improves with intravenous administration of edrophonium. Edrophonium is a fast-acting acetylcholinesterase inhibitor (AChE-I). It produces immediate and temporary relief of muscle weakness. Its onset of action is quick, starts within 30 seconds and lasts for about 5 minutes (41). On electrophysiological examination, most patients exhibit decremental electromyographic response on repetitive supramaximal stimulation of the motor nerves. The most sensitive electrodiagnostic test for MG is single-fiber electromyography, which reveals deficits of neuromuscular transmission in 95%–99% of MG patients and excludes the diagnosis of MG when it yields normal results. Increased jitter and neuromuscular blocking are seen in single fiber electromyography of patients with MG. Single-fiber electromyography selectively records action potentials from small number (usually 2 or 3) of muscle fibers innervated by a single motor unit. The amount of ACh released at the NMJ at different times has a small variability, resulting in comparable variations in the rise of end plate potentials (EPPs) and the muscle fiber pair inter-potential intervals. This variability is highly sensitive to neuromuscular transmission abnormalities and is increased in MG patients (i.e. increased jitter). Neuromuscular blocking occurs as a result of failure of transmission of one of the potentials, when one of the muscle fibers fails to transmit an action potential because EPP does not reach the necessary threshold (42).
Approximately 80%–90% of patients with generalized MG and 30-50% of patients with ocular MG are seropositive for muscle nAChRs Abs. The level of muscle nAChRs Abs considerably differs between patients, with values ranging from 0.5 nM to 1,000 nM (43). It was found that there was no correlation between the serum level of Abs and the clinical severity of the condition overtime (44). Abs to muscle-specific kinase (MuSK) have been identified in approximately 30-40% of patients with classic manifestations of MG but seronegative for muscle nAChRs Abs (45). Patients positive for anti-MuSK Abs have distinct clinical criteria which differ from classical MG. They tend to have more pronounced bulbar weakness, weakness of the neck, shoulder and respiratory muscles without ocular weakness and atrophy of the tongue and face. Also they do not respond to AChE-Is but actually worsen with these medications (46). Some seronegative patients who do not have either anti-AChR or anti-MuSK Abs might have a plasma factor that activates a second messenger pathway in the muscle, resulting in phosphorylation and inactivation of AChRs (47). MG patients may also synthesize Abs against non–muscle-specific proteins, such as myofibrillar proteins. Some Abs as anti-myosin and anti–fast troponin Abs may cross-react with AChRs (48). MuSK, agrin and rapsyn are proteins necessary for clustering of AChRs at the NMJ. MuSK is a protein present prominently at the NMJ and helps organization of AChRs on the muscle cell surface. MuSK is part of the receptor for agrin which is a protein synthesized by motor neurons and secreted into the synaptic basal lamina. Agrin/ MuSK interaction triggers and maintains rapsyn-dependent clustering of AChRs and other postsynaptic proteins. Rapsyn is a peripheral membrane protein exposed on the cytoplasmic surface of the postsynaptic membrane (49).
In adults, the thymus gland is abnormal in up to 90% of people with MG. Nearly 70% of them have enlarged thymus gland (Lymphofollicular hyperplasia), while 10-20% usually have benign thymic tumors called thymoma. In turn, it was found that 20–25% of patients with a thymoma have MG (50). It has been found that the serum from about 84-100% of young patients (onset ≤40 years) with MG and nearly 50% of patients with late-onset MG (onset ≥ 50 years) processes Abs that bind in a cross-striational pattern to skeletal and heart muscle tissue (Anti-striational Abs or anti-SM Abs). Anti-striational protein is also detectable in 30% of MG patients without thymoma (51). MG patients with thymoma have Abs against titin and ryanodine receptors (RyRs). Anti- SM Abs reacts with epitopes on the muscle protein titin and RyRs. Higher titers of anti-SM Abs are associated with more severe disease and thus can be used as a prognostic deter minant in MG (52). From the clinical perspective, the above information that the majority of patients ≤ 40 years have thymoma, highlights the importance of searching for an occult neoplasm in all patients with MG.
Till now, no therapy is found to completely eradicate MG. It is the decision for the treating neurologist to determine which treatment option is best for each individual. This decision mainly depends on the severity of the weakness, which muscles are affected and the individual’s age and other associated medical problems. The currently used treatment modalities for MG include: AChE-Is (as neostigmine and pyridostigmine) (53), immunopharmacologic drugs (as prednisone (54), azathioprine (55), cyclosporine (56), mycophenolate mofetil (57), cyclophosphamide (58), tacrolimus (59) and rituximab (60). Azathioprine, cyclosporine and Mycophenolate Mofetil are used most often in patients having relapses while on prednisone or as a steroid-sparing agent in patients taking high doses of prednisone and pyridostigmine for long periods of time. Non-pharmacologic immunotherapies include: plasma exchange (plasmapharesis) (61), intravenous immunoglobulins (IVIGs) (62) and immunoadsorption (63). Improvement with plasmapharesis occurs within few days. It is much faster than other immunomodulating, but it is temporarily and lasts from weeks to months. Plasmapheresis is an established therapy in myasthenic crisis, before thymectomy, sometimes before initiation of corticosteroids in very weak patients and for patients’ refractory to other therapies (61). High-dose IVIGs floods the body with gamma globulin antibodies from several donors (62). However, studies demonstrated that improvement is more rapid after plasmapheresis than after IVIGs (64). Immunoadsorption is capable to eliminate huge amounts of immunoglobulins from the patient’s circulation with a minimum of side effects which is in contrast to plasmapharesis which removes about 50 – 75% of antibodies and other plasmatic factors (65). This is due to the fact that immunoglobulins are distributed in the intravascular and extravascular compartments in approximately equal amounts. Inflammatory processes often occur in the tissue and not in the vascular bed. Thus simple removal of immunoglobulins from the circulation through plasmapharesis does not necessarily result in stopping the immune process. The repeated treatment cycles with adequately processed plasma volumes through immunoadsorption overcome redistribution of pathological autoantibodies (66). For critically ill patients with myasthenic crisis, the combination of AChE-Is, immunosuppressive drugs and plasma exchange has been increasingly reported. AChE-Is and immunotherapy produce marked improvement or remission in approximately 70-80% of patients (67). The mechanisms of action of different treatment modalities of MG are shown in table 2.
Because of thymus’s pathogenic role in the disease, the only useful surgical treatment in symptomatic patients is thymectomy or mediastinal dissection. The presence of a thymoma is one absolute indication for thymectomy (68). Despite the debate about the effectiveness of thymectomy in patients with MG without thymoma, several reports confirmed the effectiveness of thymectomy in early-onset MG who lacked a thymoma and for patients who do not show good response or cannot decrease the dose of medications (69). MG patients seronegative for AChR Abs (i.e. anti-MuSK Ab–positive) have shown no improvement after thymectomy because their thymi lack the germinal centers and the infiltrates of lymphocytes that characterize thymus of patients having anti-AChR Abs (70). For post-thymectomy radiotherapy; it has been indicated that thymoma with WHO cell types A, AB and B1 (71) or Masaoka stage I and II (72) do not need post-operative radiotherapy (73). In stage II thymoma, controversial views were present regarding the role of post-operative radiotherapy. However and despite an absence of a consensus, currently post-operative radiotherapy is indicated in the majority of stage II thymoma with tumors >5 cm or with radiographic evidence of invasiveness (74).
Comorbid nervous system manifestations and disorders with MG
Cognitive dysfunction with MG
Patients with MG frequently encounter memory difficulties (60%) (2,3,75) and impaired performance on variety of memory tests and measures of response fluency, information processing and verbal and visual learning (4,76), regardless to mood disturbance, disease duration, or daily dose of immunosuppressive drugs. Electroencephalographic (EEG) abnormalities (77-79) and abnormal evoked potential responses were noted in patients with MG (80-82). Abnormal immunoglobulin bands were also identified in cerebrospinal fluid (CSF) of patients with MG (83-86). Varying benefits from plasmapharesis were observed, however some studies did not report functional or reversible central deficits after treatment of MG despite improvement of muscle symptoms (4,31).
Sleep abnormalities in MG
The majority of patients with MG experience above-average number of periods of apneas and hypopnea during sleep (5-7). Both central (CSA) and obstructive (OSA) types of sleep apneas and hypopneas may occur with MG. The duration and severity of MG were found to be correlated with the severity of apneas (87). CSA describes a group of conditions in which cessations in air flow occur without respiratory effort (88). In contrast, patients with OSA have ongoing respiratory effort during respiratory events (89). CSA was reported in up to 60% of patients with MG (5) compared to 0.3%-7.8% of general population (90,91). The prevalence of OSA in patients with MG was estimated to be 36% compared to an expected prevalence of 15 to 20% in the general population. If the presence of daytime sleepiness (OSA syndrome) was included, the prevalence was 11% compared to 3% in the general population (7). During repeated episodes of OSA, breathing cessation occurs for at least 10 seconds and/or hypopnea in which an airflow reduction of at least 30% occur, are accompanied by a 4% drop in blood oxygen saturation level during sleep (89). OSA associated with MG appears to be due to bulbar fatigue or weakness and impairment of muscles of the upper airway by the disease process. This causes the diaphragm and intercostal muscles to be unable to overcome changes in airway resistance. When the patient exerts an increased effort to inspire against the occluded airway, the situation becomes worse due to the creation of more negative airway pressure. Occlusion continues until arousal occurs and the resulting increased tone of the pharyngeal muscles reopens the airway. In general, sleep apnea (whether CSA or OSA) occurs frequently during rapid eye movement sleep (REM-sleep) (92,93). This may be related to the important role of the central cholinergic system in sleep/wake rhythms and in the regulation of REM sleep, sleep perception and dreaming (89,90). While magnification of OSA during REM sleep is due to the natural loss of intercostal muscle tone during that period (94). It was found that in patients with OSA, there was an increased propensity for CSA (95).
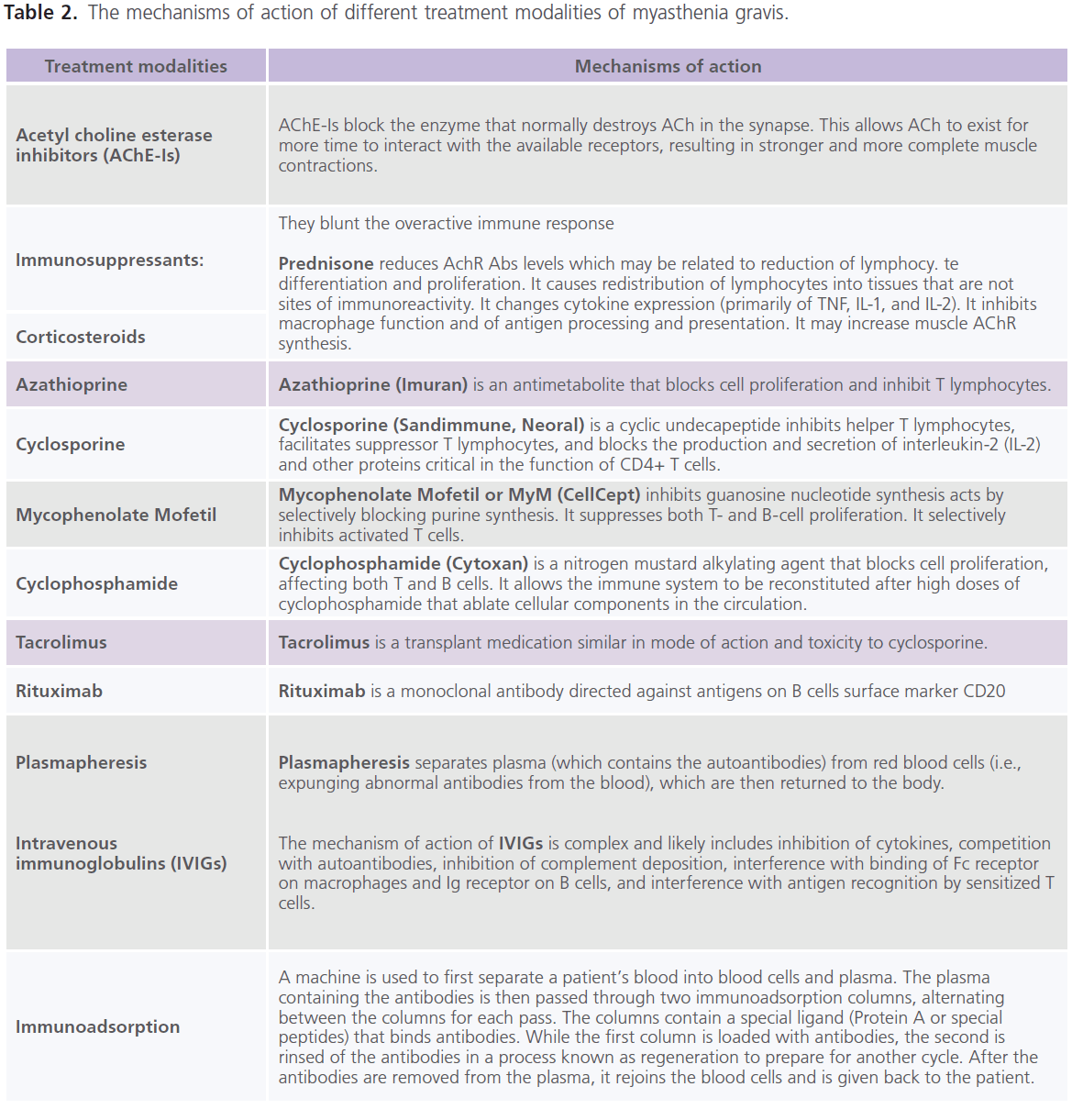
Table 2: The mechanisms of action of different treatment modalities of myasthenia gravis.
Also patients with MG may suffer from reduced sleep efficiency, reduced sleep and awakening quality, reduced REM-sleep, excessive daytime sleepiness, increased number of nocturnal awakenings, increased dream recall frequency, increased tactile sensations during dreaming and dreamed less often visually (5,96).
Autonomic and peripheral nervous system dysfunction in MG
Sympathetic and parasympathetic autonomic nervous system dysfunctions have been rarely reported with MG (8-10,97). Gastrointestinal dysmotility is a common feature in MG while isolated gastroparesis and intestinal pseudo-obstruction are rarely reported (9,98). Syncopal attacks, orthostatic hypotension, impaired heart rate variability, a low vagal tone and modified cardiac parasympathetic modulation were reported in MG (99). Acute autonomic and sensory neuropathy (AASN) and severe panautonomic failure were also reported in some cases with MG (8,9). Numbness on the limbs and loss of myelinated as well as unmyelinated fibers in biopsies of sural nerve were rarely reported with MG (8). Intestinal pseudo-obstruction, severe autonomic failure and multiple neurologic disturbances (polyneuropathy, encephalopathy, dysautonomia) occur in patients with MG usually in association with thymoma (100). Some studies reported increase in the autonomic nervous system dysfunction with the severity of MG. Also improvement of both neuromuscular and autonomic symptoms was observed with AChEI-s and after thymectomy. In contrast, others did not find significant correlations between autonomic nervous system dysfunction and disease duration, clinical manifestations, cardiovascular risk factors and diseases activity (10,98,100).
Neurophysiological testing and laboratory studies also confirm the presence of autonomic dysfunction in patients with MG. For example: 1) impaired quantitative sudomotor axon reflex test (an indicator of sympathetic deficiency) was reported in patients with MG (101), 2) augmentation in epinephrine excretion, while the nor-epinephrine excretion remains unchanged or even undergoing reduction, in response to forearm ischemia or orthostasis (a sign of sympathetic deficiency) was also reported in patients with MG (102). While in normal subjects, both stimuli induce a rise in norepinephrine urinary excretion without significant change in epinephrine excretion.
Clinical significance
The recognition of co-morbid conditions with MG is mandatory, not only for diagnosis and determining prognosis but also for patients’ management. Autonomic nervous system dysfunction is a marker for poor prognosis. MG patients with autonomic nervous system dysfunction are considered as high risk patients and are candidate for earlier considerations of cardioprotective medications, caution while prescribing drugs that disturb the cardiovascular autonomic nervous system (103) and while pre-anesthetic evaluation before surgery, for example: 1) because of muscle weakness and difficult swallowing encountered in patients with MG, awake or rapid sequence intubation is preferred and metoclopramide, H2-blockers and non-particulate antacids have to be given as prophylaxis due to the possibility of aspiration of gastric contents (104), 2) respiratory depressants, such as opioids, barbiturates, and benzodiazepines should be avoided as preanesthetic medication because some patients with MG are very sensitive to their effects (104), 3) muscular ethylephrine is commonly used before muscle blockade before thymectomy to prevent hypotension (104), 4) the intermediate or short action drugs as atracurium, rocuronium and mivacurium may be safely used as non-depolarizing neuromuscular blockers but not vecuronium as some MG patients are more sensitive to it. Succinylcholine is better avoided due to the unpredictable response from it (as resistance, prolonged effect or unexpected responses) (104-106), 5) inhalational anesthetics as isoflurane may be safely used in patients with MG as it promotes some degree of muscle relaxation and induces 30% to 50% neuromuscular block. Thiopental is better avoided for patients with MG as it may depress peripheral synapses and NMJ (107). Nitrous oxide may also be used in myasthenic patients without worsening of the disease (108). Epidural catheter using bupivacaine may be safely used in patients with MG as they allow more fractional local anesthetic doses, less hemodynamic repercussion, lower anesthetic doses and allow completion of the surgical procedure (109,110).
The possible mechanisms of comorbid nervous sytem manifestations and disorders with MG
The exact mechanism(s) of comorbid nervous system manifestations with MG is unknown. Generalized cholinergic deficiency due to involvement of nervous system cholinergic systems and pathways by the immunopathogenic process responsible for MG is highly suggested. In contrast, some authors suggested that it may be non-specific autoimmune response in presence or absence of tumor (29), or as a complication of MG (30-32).
The theory of generalized cholinergic deficiency
The theory of generalized cholinergic deficiency may be attributed to the structural identities between different muscle and neuronal nAChRs subunits with the possibility of crossreactivity between different nAChRs Abs (4,7,10,16,25,26). It may also be due to the immune responses driven by muscle and neuronal nAChRs antibodies expressed by cancer (e.g. thymoma or small cell lung cancer) (i.e. paraneoplastic syndrome) (27,28).
Structure of different nAChRs subtypes in MG and its comorbid immune mediated diseases
There are many AChR subtypes, each defined by a different combination of subunits, some of which are transiently expressed in muscle during development. Others are expressed in a wide variety of neurons, keratinocytes, vascular and bronchial epithelia (111,112).
Adult Muscle nAChR is a multimeric membrane glycoprotein consists of five subunits [two α1 and one each β, δ, and ε (epsilon) subunits], which spans the plasma membrane around a central ion channel or pore in the order α1, δ, α1, ε, β1. Each of the subunits contains an N-terminal 200-amino acid extracellular domain (113,114). When a nerve impulse travels down the nerve, the ACh neurotransmitter is released from vesicles in the nerve ending into the synapse and bathes AChRs located on the muscle side of the synaptic bouton, the depolarization opens voltage-gated Ca2+ channels on the presynaptic membrane. This Ca+2 influx triggers fusion of synaptic vesicles with the presynaptic membrane and ACh release. The ACh diffuses into the synaptic cleft and reaches and binds to AChRs, thereby triggering the opening of their cation channels and influx of Na+ into the muscle fiber. The resulting EPP activates voltage-gated Na+ channels, leading to further influx of Na+ and spreading of the action potential along the muscle fiber. The reaction is short-lived; as within a very brief time, the ACh in the receptors is metabolized into its components (acetate and choline) by the acetylcholine esterase (AChE) enzyme. Then any remaining ACh diffuses away from the receptors.
The neuronal nAChRs are made up of various combinations of α (α2–α10) and β (β1–β4) subunits. Neuronal subunits are positioned in widespread CNS locations including cortex, hippocampus, midbrain, and brainstem (115-117). When ACh binds to the specific neuronal α2 subunits, the ion channel opens transiently causing transient membrane depolarization. The primary autonomic ganglion AChR subtype (ganglionic nAChRs) are pentameric ligand-gated cation channels containing two α3 and β4 subunits, but β2 or α5, α7 subunits may also be associated. The ganglionic nAChRs mediate fast neurotransmission at synapses in the mammalian sympathetic and parasympathetic nervous system and enteric autonomic ganglia. The α9 subunit of neuronal nAChR is originally discovered in the cochlea and sensory ganglia of the nervous system (118-120). The brain dopaminergic and adrenergic neurons contain minor nAChRs as α6 subunits in combination with β2, β3, and often α3 or α4 subunits. These subunits contribute to binding sites for ACh and the agonist epibatidine (121,122). The α9 is one of the neuronal AChRs capable of binding α-bungarotoxin (BTx) (like α1 and α7) (123).
Due to diverse functional roles of nAChRs, antibody-mediated autoimmune response to different subunits may be presented by various neurologic and medical problems as follow: 1) antibodies against the two α1-subunits type of muscle nAChRs were identified in patients with MG and experimental autoimmune MG (EAMG) (1,124), 2) antibodies against α7-type of neuronal nAChRs were identified in few patients with encephalitis (125), 3) antibodies against α3 subunit of ganglionic nAChR antibodies were identified in up to 50% of patients with autoimmune autonomic neuropathy (AAN) (also known as subacute pandysautonomia) (126,127) and experimental autoimmune autonomic neuropathy (EAAN) (128), and 4) antibodies against the α9 of nAChRs were identified with an autoimmune disorder pemphigus vulgaris (129).
Structural identities between different nAChRs subunits with the possibility of cross-reactivity between anti-nAChR α1 antibodies and other nAChR subunits
Different nAChRs subunits share some structural features with differing amounts of identity among various α subunits. The identity rates have been found to range between 35% and 73%. Immediately following the large extracellular domain in all AChR subunits, there are three closely spaced transmembrane domains, M1-M3. The three domains (M1- M3) comprise about 90 conserved amino acids. M1 links se quences in the extracellular domain that contributes to the ACh-binding site with a short sequence near the cytoplasmic surface between M1 and M2 that forms the channel gate (113,130). Some rat muscle antibodies to the main immunogenic region (MIR) on α1 react very well with human α3 and α5 subunits. A disulfide linked-loop corresponding to amino acids 128-142 of α1 subunits is characteristic of all subunits in the superfamily. The α1, α3, α5, β3 subunits are quite similar in their sequences in the 66-76 region (26). There is considerable identity between muscular α1 and neuronal α3 and α9 subunits. The extracellular domain of α 3 subunits in the ganglionic AChR is 60% identical in amino acid sequence of the muscle α 1 subunit (131). The identity between α1 and α9 for the whole length of the molecules is 25% and 37.5% or up to 43% (132).
The structural similarity with α1-subunit might be enough for the cross-reaction of their antibodies and anti-α3 and anti-α9 antibodies. This supports the possibility that AChR α3 and α9 subunits may be the target autoantigen in MG associated CNS and autonomic symptoms and the antigenic modulation and complement attacks operate in decreasing the ACh analogous in other body systems similar to that for the NMJ (113,130). However, it is even conceivable that an immune response to different nAChRs α subunits (α1, α3, α9) could result in different clinical manifestations in different individuals.
suggestion that cross-reactivity between anti-AChR α1 antibodies and other nAChR subunits as a cause of generalized cholinergic deficiency resulting in additional nervous system manifestations and disorders in patients with MG, is further supported by the following observations: 1) nAChR Abs other than muscle nAChRs Abs were detected in patients with MG. For example: Abs to ganglioside or α3 subunit of nAChRs were also reported in of patients with idiopathic peripheral neuropathy or idiopathic dysautonomia, and celiac disease (133). Seropositivity rate for ganglionic AChR antibodies was found to be 50% in some patients with MG (9,10,97). Abs to ganglionic AChRs were reported in the serum and CSF of the majority of patients with MG and subacute autonomic neuropathy (10). Abs to α9 subunit of nAChRs were identified in the serum samples of mice with EAMG (134), 2) It was observed that intraventricular administration of MG patients’ sera exerted adverse effects on cerebral evoked potentials and hypothalamic functions (135), 3) Electrophysiological features of postsynaptic failure of ACh transmission was observed in autonomic ganglia from α3 AChR–immunized rabbits (127,128,136), 4) Ganglionic AChR IgG was found to inhibit nicotinic membrane current in cultured neuroblastoma cells (137), 5) Mutations in some nAChRs subunits was found to be associated with rare forms of epilepsy and dysautonomia (138,139), and 6) The involvement of the central nAChRs and central cholinergic pathways by the disease process cannot be denied as a cause of central deficits (e.g. cognitive and sleep abnormalities) observed in patients with MG. The hippocampus, a cerebral structure highly involved in learning and memory, is a target for abundant cholinergic innervation. Hippocampal nAChRs can modulate synaptic plasticity via mechanisms involved in long-term potentiation (LTP) (140). This is further supported by the finding that AChE-Is may improve memory functioning in diverse neurological conditions associated with memory deficits (e.g. Alzheimer’s disease, Parkinson’s disease, etc) (141-143). Because of the role of cholinergic function in memory and related cognitive processes and sleep (92-94), dysfunction of the central cholinergic system has been accused to underlie the cognitive deficits, sleep disorders and EEG abnormalities observed in patients with MG.
Immunopathogenesis of MG and tumor immunology
Immunopathogenesis of MG
In both MG and EAMG, the antibodies are bound to AChRs at the postsynaptic membrane. EAMG, the animal model of MG, is produced in rabbits and rodents by immunization with muscle-type nAChR protein. EAMG closely mimics MG in its clinical and immunopathological manifestations (124). MG and EAMG are T cell-dependent and antibody-mediated autoimmune diseases (B-cell mediated disease) (144). The relationship between the thymus gland and MG is well established. The thymus gland plays an important role in the development of the immune system. The thymus is the central organ in T cell–mediated immunity. The myoid cells (musclelike cells) in the thymus might be responsible for the autoimmune reaction seen in MG. T cells are first sensitized against myoid cells within the thymus and causes the formation of germinal centers, which are key facilitators in the autoimmune reaction against the body’s nAChRs (144,145). AChRspecific CD4+ T cells with T helper function in the blood and thymus are necessary for the development of MG symptoms. In MG and EAMG, T helper (Th) lymphocytes and cytokines are important for immunoregulation. Naive CD4+ Th0 cells (antigen-inexperienced) are bipotential cells. Cytokines present in the microenvironment and produced by the cells of the innate immune system are important factors that influence the differentiation of T0 cells toward the Th1 or Th2 subsets. Differentiated CD4+ T cells are classified into subtypes based on the cytokines they secrete. Th1 cells secrete IL-12, IFN-γ, and TNF-α proinflammatory cytokines which are important in cell-mediated immune responses. Proinflammatory Th1 cytokines induce expression of MHC class II molecules in muscle, thereby facilitating presentation of muscle AChR epitopes and further expansion of activated anti-AChR CD4+ T cells. IL-12 secreted by Th1 cells is a crucial cytokine for differen tiation of Th1 cells which is necessary for development of EAMG. Moreover, it has been found that estrogen enhances EAMG development in mice by promoting augmented IL-12 production by AChR-specific Th1 cells, suggesting that estrogens mediate sex differences in autoimmunity because of a Th1-mediated mechanism (146). Increased IFN-γ production may explain the increased expression of IFN-γ–induced chemokines and monokines and their receptors in muscle, thymus, and lymph nodes in MG patients and rats with EAMG. Th2 cells secrete IL-4, IL-5, IL-6 and IL-10 anti-inflammatory cytokines which are also important inducers of humoral immune responses. IL-5, IL-6, and IL-10 are important for the development of EAMG. IL-10 secreted by Th2 cells is a potent growth and differentiation factor for B cells which facilitates the development of EAMG and human MG. In contrast, IL-4 secreted by Th2 cells appears to be involved in the differentiation of AChR-specific regulatory CD4+ T cells, which can prevent the development of EAMG and its progression to a self-maintaining. However, there is no direct evidence for the role of IL-4 in immuno-modulation in human MG (147). Furthermore, IL-4 stimulates differentiation of Th3 cells, which secrete TGF-β and are involved in immunosuppressive mechanisms (148).
It has been suggested that autonomic manifestations in MG as the low noradrenergic-activity and high adrenergic-activity levels during the basal (supine-resting) state, as well as after several stress tests (e.g. exercise) are in favor of Th-1 immunosuppression plus Th-2 predominance (149). β2 adrenergic receptors are expressed on various types of immune cells including T, B and antigen presenting cells (APCs) (150). In autoimmune diseases (150), pro-inflammatory cytokines are produced during the Th1 type immune responses including TNF-α, IFN-γ, IL-12, IL-1β and IL-6 can inhibit local sympathetic tone, whereas they increase systemic sympathetic tone due to the counter-regulatory effects of Th1/Th2 type immune responses, Th2 immune-deviation decreases systemic sympathetic tone (151).
The balance of antigen-specific Th1/Th2 cells may dictate the clinical outcome of an immune system related disease. Activated CD4+ T cells interact with B cells (which secretes) low-affinity anti-nAChR Abs. This triggers somatic mutations of the Ig genes, leading to synthesis of high-affinity Abs (IgGs) (pathogenic anti-nAChR Abs). An augmented production of autoreactive CD4+ cells, on one side and an increase of the immunoregulatory T cells that augment autoantibody production, on the other side may have a significant role in sustaining the immune response in EAMG and reflects the increased entry of activated autoreactive CD4+ T cells from the periphery into the thymus. Similar putative mechanisms may be suggested to underlie sustained autoimmune response in human MG (152). Other CD4+ T cell subtypes may have a role in MG. CD4+ T cells that express the CD25 marker and the transcription factor Foxp3 are known as Tregs and are important in maintaining self-tolerance. Tregs in MG patients may be functionally impaired (153). In addition, the number of circulating Tregs has been shown to increase after thymectomy and the increase correlated with symptom improvement (154).
Antibody response in MG is polyclonal. In an individual patient, antibodies are composed of different subclasses of IgG. More than 50% of Abs binds to the extracellular domains of the two α1-subunits of the muscle nAChRs at a site called the MIR (44). Binding of Abs to the muscle nAChRs at the NMJ will affect the neuromuscular transmission by at least 3 mechanisms: a) activation of complement at the NMJ, b) accelerated degradation of AChRs cross-linked by Abs, and c) functional AChRs block. Complement activation at the NMJ might be the primary cause of AChRs loss and failure of neuromuscular transmission. The complements C3, C9, and C5-9 activation neoantigens are thought to potentiate degeneration of AChRs and post-synaptic membrane lysis (155). It has been found that endogenous molecules as decay-accelerating factor (DAF or CD55), the membrane cofactor protein (MCP or CD46), and the membrane inhibitor of reactive lysis (MIRL or CD59) act as intrinsic complement regulators and are capable to protect membrane cells from activation by autologous complement on their surfaces (156-158). In human MG and EAMG, Th1 CD4+ cells drive the synthesis of anti-AChR complement-fixing IgG subclasses. The modulation of AChRs by IgG-complement interaction accelerates the internalization of AChRs and accelerates degradation of AChRs cross linked by Abs (a process known as antigenic modulation). Antigenic modulation is the ability of an Ab to cross-link 2 antigen molecules, thereby triggering a cellular signal that causes accelerated endocytosis, shedding of the AChRs into the synaptic space and degradation of the cross-linked molecules. If accelerated degradation is not compensated by increased AChRs synthesis, it will lead to a reduction of the available AChR molecules at the NMJ and myasthenic symptoms. The net result is destruction of segments of the post-synaptic membrane and disruption of its architecture. The decrease in the density of AChRs and the restriction of the membrane surface available for the insertion of new AChRs cause reduction of the amount of depolarization at the NMJ and inability to trigger muscle activity (2). However, not all anti-AChR Abs cause antigenic modulation because, even though all IgG Abs have 2 antigen-binding sites, the epitope location on the AChR surface may restrict the ability of Abs to cross-link a second AChR molecule (159). Also many MG patients have low levels of anti-AChR Abs that recognize the ACh-binding site; these might block the AChR in spite of their low concentration and contribute to acute, severe muscle weakness and myasthenic crises without either inflammation or necrosis of the NMJ. Functional AChR block is due to Ab binding to the ACh-binding site is an uncommon pathogenic mechanism in MG, but it may be clinically important (160).
Till now, there is lack of knowledge of the exact culprit antigen(s) responsible for MG and its comorbid disorders, thus advances in understanding the cellular and molecular mechanisms involved in neuro-immune pathways of MG may result in proper antigen-specific therapeutic strategies for total eradication of the disease without affecting other functions of the immune system or causing adverse effects. Several new approaches have proven to be successful in EAMG which include: a) administration of AChR or parts of its sequence in a manner known to induce tolerance: it has been suggested that experimental antigen presentation under special circumstances may lead to antigen-specific tolerance rather than activated CD4+ T cells. Recently, several studies have demonstrated that treatment of dendritic cells (DCs) with TGF-β, IFN-γ, or IL-10 before injection in rat model of EAMG, suppressed or ameliorated the myasthenic symptoms (148,161,162). This effect has been attributed to the reduced production of anti-AChR Abs without a reduced proliferative response of T cells to the AChR. Approaches which use tolerance-inducing antigen presenting cells (APCs), which present all AChR epitopes might be useful for the treatment of MG as it influences all AChR-specific T cells, b) T cell vaccination: T cell vaccination is already used in clinical trials for the treatment of multiple sclerosis, rheumatoid arthritis, and psoriasis. It is effective in EAMG, and it is a promising future strategy for the treatment of MG. The mechanisms of action of T cell vaccination are complex, and they likely include the induction of modulatory CD4+ and CD8+ T cells (163). Another approach used synthetic peptide analogs of an epitope recognized by autoimmune CD4+ T cells that bind the MHC class II molecules but cannot stimulate the specific CD4+ cells. These are known as altered peptide ligands (APLs). APLs compete with peptide epitopes derived from the autoantigen, thereby turning off the autoimmune response. APLs might also stimulate modulatory anti-inflammatory CD4+ T cells or anergize the pathogenic CD4+ T cells (164), c) Interference with formation of the complex between MHC class II molecules, epitope peptide, T cell receptor and CD4 molecule: Blocking complement component 6 was found to be effective in rodent EAMG and protect rodents from EAMG CR1 Soluble recombinant receptor that competitively inhibits complement (165), d) Long-term therapeutic use of EN101, an antisense oligonucleotide that suppresses the expression of AChE-R: This new approach is based on the observation that in MG and EAMG animals, there is increase in the rate of ACh hydrolysis and the efficacy of AChR activation. This is caused by enhanced transcription and accumulation of the rare read through AChE-R variant instead of the commonly occurring synaptic AChE-S. The commonly occurringsynaptic AChE-S variant forms membrane multimers while AChE-R soluble monomers that lack the carboxyterminal cysteine needed for membrane attachment. AChE-R permeates the synaptic space and degrades ACh before it reaches the postsynaptic membrane, thereby compromising AChR activation. EN101 has been suggested to normalize neuromuscular transmission in EAMG by modulating the synthesis of AChE variants (166,167).
Tumor immunology and MG and its related immune-mediated comorbidities
NMJ disorders are observed in association with certain tumors as paraneoplastic neurological manifestations. The immune responses driven by muscle and neuronal nAChR subtypes expressed in cancer may account for several related paraneoplastic neurological disorders affecting the cholinergic systems. MG is frequently associated with WHO type B1 and B2 thymomas (71). A broad spectrum of neurological and non-neurological paraneoplastic diseases was observed in association with MG either in the presence of the thymoma or at different times after thymomectomy, which include: Lambert- Eaton mysthenic syndrome (LEMS) (168), AAN, neuromyotonia, limbic encephalopathy, encephalomyelitis, seizures, dementia, movement disorders, cerebellar degeneration, subacute hearing loss, psychosis and sleep disorders (28). Nonneurological paraneoplastic diseases include: hematological and cutaneous diseases prevailed as pemphigus vulgaris (169), diffuse alopecia areata and pemphigus foliaceus (170).
LEMS is caused by Abs against neuronal P/Q-type voltagegated calcium channels (VGCC) that are present in the presynaptic somatic motor terminals and parasympathetic effector junctions but they do not act via complement-mediated lysis (171-173). Abs appear to act only by cross-linking the VGCCs on the motor nerve terminal surface and causing their clustering and internalization. This causes reduction in the number of VGCCs, reduction in the calcium-induced release of ACh and thus reduction in AChR activation. LEMS is characterized by dysautonomia, mostly involving the parasympathetic system, erectile dysfunction, hypohidrosis (dry eyes and mouth), constipation and abnormal papillary responses to light and accommodation which are common autonomic manifestations in LEMS and quite distinct from the severe dysautonomia and gastrointestinal dysmotility encountered with MG (9). In some patients with thymoma, MG-LEMS was reported with evidence of antibody activity directed against postsynaptic AChRs, presynaptic somatic motor terminals, and autonomic effector junctions in patients with malignant thymoma and complete remission followed surgery (25). The interaction impairs the ACh release leading to weakness and autonomic dysfunction. AAN is caused by antibodies against neuronal nAChRs in the autonomic ganglia (174). It is characterized by subacute onset gastrointestinal dysmotility with pain and vomiting, dry eyes and mouth (a combination of sicca complex), abnormal papillary responses to light and accommodation, impaired heart rate variability, orthostatic hypotension and up to complete pandysautonomia. Neuromyotonia is a peripheral ion channel disorder due to presence of antibodies to voltage-gated potassium channels (VGKCs). Patients with neuromyotonia complain of muscle cramps and twitching due to reduction of the number of VGKCs at the motor nerve terminals and motor nerves itself by antibodies rendering the nerve membrane potential unstable and hyperexcitable (175).
There is also a recognized link between ectopic expression of neuronal and muscle autoantigens in certain cancers other than thymoma, for example: small cell lung cancer (SCLC), and rarely ovarian, breast and thyroid tumors and Hodgkin’s lymphoma (176,177). Several neuronal nAChR subtypes, as well as muscle nAChR, have previously been found in SCLC cell lines. Functional studies in SCLC cell lines suggest that these ligand-gated cation channels may act in synergy with VGCC to influence transcription of growth-regulatory genes (176). They also regulate cell proliferation and secretion of autocrine growth factors (178,179). Approximately 50% of patients with LEMS have SCLC (177). Other paraneoplastic diseases observed with SCLC include: limbic encephalopathy, encephalomyelitis, seizures, dementia, movement disorders, cerebellar degeneration, subacute hearing loss, psychosis and sleep disorders (28).
Some authors suggested that the associated neoplasm may present multiple antigens that trigger several autoimmune responses. In some patients with MG and thymoma, a high prevalence of glutamic acid decarboxylase-65 (GAD-65) Abs and Ab titers were identified and found to be correlated with severity of associated parasympathetic nervous system, gastrointestinal dysmotility and autonomic dysfunction (180). Paraneoplastic autonomic disorders are associated with various overlapping antibody associations, including GAD-65 (glutamic acid decarboxylase-65) (180), anti-Hu (ANNA-1) (181,182), ganglionic AChRs (183), CRMP-5 (184), and PCA- 2 (185). In general for the paraneoplastic syndrome, both T and B cells have a role in its pathogenesis. T cells have the potential to recognize tumor cells and to cross into the brain or the peripheral nervous system to attack neurons. T cells induce expression of MCH class I molecules by neurons, thus, increasing their susceptibility to become targets for T cell attack (186). Activated, plasma cells produce autoantibodies directed against tumor antigens. They differentiate in situ from circulating B cells that have traveled to the brain or peripheral nervous system (187). There is evidence that neurons can take-up extracellular molecules such as Abs and also, paraneoplastic syndrome Abs are able to infiltrate neurons. Neuronal antigens may trigger apoptotic neuron death by interfering with essential intracellular processes. This process would result in the production of further Abs that could accumulate at high titers in both the blood and CSF and further amplify the autoimmune response. Advances in the investigation on immunological function of thymoma helps to elucidate the pathogenesis of thymoma-related autoimmunity and the high affinity of thymoma with MG. This association is thought to depend on thymoma’s capacity to produce and export autoreactive T cells causing autoimmunity (183,188). Thymomas produce significant number of CD4+CD8+ double-positive T cells, and at the same time, the neoplastic epithelial cells express HLA-DR molecules at a slightly reduced level compared with the normal thymus. The impaired expression of HLA-DR molecules in neoplastic epithelial cells of thymomas possibly affects positive selection of CD4+CD8− single-positive T cells and may result in alteration of its repertoire (188).
Non-specifically acting mechanisms and consequences of MG
Multiple non-specific autoimmune response:
Some authors suggested that the co-morbid neurological and non-neurological manifestations and disorders might reflect a non-specific multiple autoimmune response or response to un-specifically acting cytokines in presence or absence of tumor (markers of autoimmunity). In support: 1) MG was reported in association with other non-nervous system medical immune-mediated disorders as diabetes mellitus (189), celiac disease (133), thyroiditis (190), vitiligo (191), atopic dermatitis (192), rheumatoid arthritis (193), systemic lupus erythematosis (194), polyarteritis nodosa (195), Crohn’s disease (193) and idiopathic thrombocytopenic purpura (196). 2) Other example is that half of the acquired severe cases of autonomic neuropathies were found to be associated with celiac disease and systemic lupus erythematosus, Isaacs syndrome, LEMS, dementia, sensory neuropathy, diabetes, amyloidosis, drugs, toxins or idiopathic and high titers of antibody to ganglionic nAChRs (α3 AChR) (133).
Complications of MG and its medications:
Because of the very low concentrations of muscle nAChR antibodies in the CSF of patients with MG, some authors suggested that it is highly unlikely that the CNS cholinergic systems are affected by muscle antibodies and that the co-morbid manifestations and disorders might reflect associated peripheral mechanisms as mood disorders, hypoxia or as an adverse effect from AChEI-s used to treat MG (30-32). Enhanced gastrointestinal motility is a common side effect of AChE-Is as they increase the cholinergic activity of parasympathetic ganglia innervating the gut (197). Some suggested that the respiratory impairment (apnea/hypop nea) and hypoxia caused by oropharyngeal, intercostal and diaphragmatic muscle weakness which worsen during REM sleep might be responsible for the sleep-wake cycle abnormalities and EEG abnormalities noted in patients with MG (93). Micro-arousals may occur whenever an apneic or hypopnic event and disrupts sleep. Sleep fragmentation and lack of REM sleep and severely disrupted sleep patterns may result in personality changes, including irritability, anxiety, depression and decreased sexual drive or impotence (198). The encountered sleep disturbances can also impair memory and performance of MG patients on neuropsychological tests (5). It is also possible that the resulting oxygen desaturation which occur during long apneic events (last for 2 minutes or longer) may also be related to cognitive dysfunction in patients with MG (199). This is supported by the findings that: the sleep apneas found in patients with MG have been found to be strongly related to periods of confusion, the inability to concentrate, shortened attention span throughout the waking hours, or short-term memory impairment (5). Some studies indicate that physical fatigue, an important symptom of MG, produces cognitive fatigue, memory impairment and pervasive impairments in important aspects of patients’ lives. This was based on the observation that the patients’ perceptions of both cognitive and physical fatigue have been observed to be increased significantly following completion of demanding cognitive work and this phenomena was not observed in control subjects (32).
Conclusions
Some patients with MG may experience comorbid nervous system manifestations and syndromes as memory difficulties, sleep abnormalities, autonomic dysfunction, peripheral neuropathy, epilepsy and psychiatric disorders. Recognition of co-morbidities with MG is mandatory for diagnosis, determining prognosis and management. The exact mechanisms of such comorbidities are unknown, however they may be due to the involvement of nervous system cholinergic systems and pathways by the immune mediated process of MG, paraneoplastic response to certain tumor antigens or response to non-specific autoimmune process or less likely as a complication of MG and its medications. Future advances in understanding the molecular and cellular mechanisms of MG may provide information regarding the exact mechanisms MG and its comorbidities. They may also provide satisfactory answers to some challenging questions, for example: a) why an immune response to different nAChRs α subunits (α1, α3, α9) could result in different clinical manifestations in different individuals?, b) why some patients with MG may develop medical or non-neurological autoimmune disease?, c) why does improvement after treatment occur in some receptors (i.e. muscle) and not others (e.g. neurons)?, d) why some patients develop nervous system manifestations after thymectomy?.
Conflicts of interest
None
6459
References
- Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Neurology 1976;26(11):1054–1059.
- Tucker DM, Roeltgen DP, Wann PD, Wertheimer RI. Memory dysfunction in myasthenia gravis: Evidence for central cholinergic effects. Neurology 1988;38(3):1173–1177.
- Riker WF. Memory impairment in myasthenia gravis. Neurology 1989;39(4):611-612.
- Glennerster A, Palace J, Warburton D, Oxbury S, Newsom-Davis J. Memory in myasthenia gravis: neuropsychological tests of central cholinergic function before and after effective immunologic treatment. Neurology 1996;46(4):1138-1142.
- Stepansky R, Weber G, Zeitlhofer J. Sleep apnea in myasthenia gravis. Wien Med Wochenschr 1996;146(9-10):209-210.
- Amino A, Shiozawa Z, Nagasaka T, Shindo K, Ohashi K, Tsunoda S, Shintani S. Sleep apnoea in well-controlled myasthenia gravis and the effect of thymectomy. J Neurology 1998;22(2):77-80.
- Nicolle MW, Rask S, Koopman WJ, George CF, Adams J, Wiebe S. Sleep apnea in patients with myasthenia gravis. Neurology 2006;67(1):140-142.
- Senda Y, Sugimura K, Koike Y, Matsuoka Y, Takahashi A. Concurrence of acute autonomic and sensory neuropathy and myasthenia gravis- -a case report and pathogenetic considerations. Rinsho Shinkeigaku 1989;29(3):332-325.
- Pande R, Leis AA. Myasthenia gravis, thymoma, intestinal pseudoobstruction, and neuronal nicotinic acetylcholine receptor antibody. Muscle Nerve 1999;22(11):1600-1602.
- Vernino S, Cheshire WP, Lennon VA. Myasthenia gravis with autoimmune autonomic neuropathy. Auton Neurosci 2001;88(3):187- 192.
- Grira M, Benammou S, Lamouchi T, Harzallah MS, Benslamia L. Myasthenia gravis and Epilepsy and myasthenia: a case report. Rev Neurol (Paris) 2004;160(1):93-95.
- Kwan SY, Lin JH, Su MS. Coexistence of epilepsy, myasthenia gravis and psoriasis vulgaris. Zhonghua Yi Xue Za Zhi (Taipei) 2000;63(2):153- 157.
- Low PA, Vernino S, Suarez G. Autonomic dysfunction in peripheral nerve disease. Muscle Nerve 2003;27(6):646-661.
- Somer H, Muller K, Kinnunen E. Myasthenia gravis associated with multiple sclerosis. Epidemiological survey and immunological findings. J Neurol Sci 1989;89(1):37-38.
- Isbister CM, Mackenzie PJ, Anderson D, Wade NK, Oger J. Cooccurrence of multiple sclerosis and myasthenia gravis in British Columbia. Mult Scler 2003;9(6):550-553.
- Gotkine M, Fellig Y, Abramsky O. Occurrence of CNS demyelinating disease in patients with myasthenia gravis. Neurology 2006;67(5):881- 883.
- Basiri K, Etemadifar M, Maghzi AH, Zarghami N. Frequency of myasthenia gravis in multiple sclerosis: Report of five cases from Isfahan, Iran. Neurol India 2009;57(5):638-640.
- van de Warrenburg BP, Hengstman GJ, Vos PE, Boerman RH, ter Laak HJ, van Engelen BG. Concomitant dermatomyositis and myasthenia gravis presenting with respiratory insufficiency. Muscle Nerve 2002;25(2):293-296.
- Furukawa Y, Yoshikawa H, Yachie A, Yamada M. Neuromyelitis optica associated with myasthenia gravis: Characteristic phenotype in Japanese population. Eur J Neurol 2006;13(6):655-658.
- McKeon A, Lennon VA, Jacob A, Matiello M, Lucchinetti CF, Kale N, Chan KH, Weinshenker BG, Apiwattinakul M, Wingerchuk DM, Pittock SJ. Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 2009;39(1):87-90.
- Colomb-Lippa D, King-Schumacher M, Klingler AM. Quick recertification series: Alzheimer’s disease and myasthenia gravis. JAAPA 2009;22(7):48,50.
- Ananth J, Davies R, Kerner B. Psychosis associated with thymoma. J Nerv Ment Dis 1984;172(9):556-558.
- Magni G, Micaglio GF, Lalli R, Bejato L, Candeago MR, Merskey H, Angelini C. Psychiatric disturbances associated with myasthenia gravis. Acta Psychiatr Scand 1988;77(4):443–445.
- Rohr W. Obsessive behavior and thoughts in patients with myasthenia gravis. Schweiz Arch Neurol Psychiatr 1992;143(2):105-115.
- Tabbaa MA, Leshner RT, Campbell WW. Malignant thymoma with dysautonomia and disordered neuromuscular transmission. Arch Neurol 1986;43(9):955-957.
- Lindstrom JM. Nicotinic acetylcholine receptors of muscles and nerves: comparison of their structures, functional roles, and vulnerability to pathology. Ann N Y Acad Sci 2003;998:41-52.
- Darnell RB. Paraneoplastic neurological disorders: windows into neuronal function and tumor immunity. Arch Neurol 2004;61(1):30– 32.
- Evoli A, Minicuci GM, Vitaliani R, Battaglia A, Della Marca G; Lauriola L, Fattorossi A. Paraneoplastic diseases associated with thymoma. J Neurol 2007; 254(6):756-762
- Thorlacius S, Aarli JA, Riise T, Matre R, Johnsen HJ. Associated disorders in myasthenia gravis: autoimmune diseases and their relation to thymectomy. Acta Neurologica Scandinavica 1989;80(4):290-295.
- Lewis SW, Ron MA, Newsom-Davis J. Absence of central functional cholinergic deficits in myasthenia gravis. J Neurol Neurosurg Psychiatry 1989;52(2):258–261.
- Paul RH, Cohen RA, Zawacki T, Gilchrist JM, Aloia MS. What have we learned about cognition in myasthenia gravis?: a review of methods and results. Neurosci Biobehav Rev 2001;25(1):75-81.
- Paul RH, Cohen RA, Gilchrist JM. Ratings of subjective mental fatigue relate to cognitive performance in patients with myasthenia gravis. J Clin Neurosci 2002;9(3):243-246.
- Kurtzke JK, Kurland LT. Epidemiology of neurologic diseases; in baker AB, Baker LH (eds): Clinical Neurology. Philadelphia, Harper and Row 1982;47-49.
- Sorensen T, Holm EB. Myasthenia gravis in the country of Viborg, Denmark. Eur Neurol 1989;29(3):177-179.
- Phillips LH 2nd. The epidemiology of myasthenia gravis. Neurol Clin 1994;12(2):263-271.
- Drachman DB. Myasthenia gravis. N Engl J Med 1994;330(25):1797– 1810.
- Osserman KE,ed. Myasthenia gravis. New York: Grune and Stratton 1958; 80.
- Jaretzki A 3rd, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, Sanders DB. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Ann Thorac Surg. 2000 Jul;70(1):327-334.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann N Y Acad Sci 1981;377:670-677.
- Engel AG. Disturbances in neuromuscular transmission; in Engel AG, Franzini-Armstrong C (eds): Myology. McGraw-Hill, 2nd edition, 1994;2:1769-1797.
- Boruchoff SA, Goldberg B. Edrophonium (tensilon) in diagnosis of ocular myasthenia gravis. AMA Arch Ophthalmol 1955;53(5):718-719.
- Howard JF Jr, Sanders DB, Massey JM. The electrodiagnosis of Myasthenia gravis and the Lambert Eaton myasthenic syndrome. Neurol Clin 1994;12(2):305-330.
- Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science 1973;180(88):871–872.
- Lennon VA, Griesmann GE. Evidence against acetylcholine receptor having a main immunogenic region as target for autoantibodies in myasthenia gravis. Neurology 1989;39(8):1069–1076.
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 2001;7(3):365–368.
- Pasnoor M, Wolfe GI, Nations S, Trivedi J, Barohn RJ, Herbelin L, McVey A, Dimachkie M, Kissel J, Walsh R, Amato A, Mozaffar T, Hungs M, Chui L, Goldstein J, Novella S, Burns T, Phillips L, Claussen G, Young A, Bertorini T, Oh S. Clinical findings in MuSK-antibody positive myasthenia gravis: a U.S. experience. Muscle Nerve 2010;41(3):370- 374.
- Plested CP, Tang T, Spreadbury I, Littleton ET, Kishore U, Vincent A. AChR phosphorylation and indirect inhibition of AChR function in seronegative MG. Neurology 2002;59(11):1672–1673.
- Mohan S, Barohn RJ, Jackson CE, Krolick KA. Evaluation of myosinreactive antibodies from a panel of myasthenia gravis patients. Clin Immunol Immunopathol 1994;70(3):266–273.
- Boneva N, Frenkian-Cuvelier M, Bidault J, Brenner T, Berrih-Aknin S. Major pathogenic effects of anti-MuSK antibodies in myasthenia gravis. J Neuroimmunol 2006;177(1-2):119–131.
- Onodera H. The role of the thymus in the pathogenesis of myasthenia gravis. Tohoku J Exp Med 2005;207(2):87-98.
- Romi F, Skeie GO, Gilhus NE. Striational antibodies in myasthenia gravis: reactivity and possible clinical significance. Arch Neurol 2005;62(3):442-446.
- Baggi F, Andreetta F, Antozzi C, Simoncini O, Confalonieri P, Labeit S, Cornelio F, Mantegazza R. Anti-titin and antiryanodine receptor antibodies in myasthenia gravis patients with thymoma. Ann N Y Acad Sci 1998;841:538–541.
- Scheife RT, Hills JR, Munsat TL. Myasthenia gravis: signs, symptoms, diagnosis, immunology, and current therapy. Pharmacotherapy 1981;1(1):39-54.
- Bedlack RS, Sanders DB. Steroid treatment for myasthenia gravis: steroids have an important role. Muscle Nerve 2002;25(1):117–121.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998;50(6):1778–1783.
- Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci 1993;681:539–551.
- Chaudhry V, Cornblath DR, Griffin JW, O’Brien R, Drachman DB. Mycophenolate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology 2001;56(1):94–96.
- Drachman DB, Jones RJ, Brodsky RA. Treatment of refractory myasthenia: “rebooting” with high-dose cyclophosphamide. Ann Neurol 2003;53(1):29–34.
- Ponseti JM, Azem J, Fort JM, Codina A, Montoro JB, Armengol M. Benefits of FK506 (tacrolimus) for residual, cyclosporin- and prednisone resistant myasthenia gravis: one-year follow-up of an open-label study. Clin Neurol Neurosurg 2005;107(3):187–190.
- Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant 2006;6(5 Pt 1):859–866.
- Chiu HC, Chen WH, Yeh JH. The six year experience of plasmapheresis in patients with myasthenia gravis. Ther Apher 2000;4(4):291-295.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002;105(1):3-8.
- Psaridi-Linardaki L, Trakas N, Mamalaki A, Tzartos SJ. Specific immunoadsorption of the autoantibodies from myasthenic patients using the extracellular domain of the human muscle acetylcholine receptor alpha-subunit. Development of an antigen-specific therapeutic strategy. J Neuroimmunol 2005;159(1-2):183–191.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001;25(12):967-973.
- Keller F, Wagner K, Faber U, Scholle J, Neumayer HH, Maiga M, Schultze G, Offermann G, Molzahn M: Elimination kinetics of plasma exchange. Klin.Wochenschr. 1983;61(22):1115-1122.
- Braun N, Gutenberger S, Erley CM, Risler T: Immunoglobulin and circulating immune complex kinetics during immunoadsorption onto protein A sepharose. Transfus.Sci. 1998;19 (Suppl.):25-31.
- Berrouschot J, Baumann I, Kalischewski P, Sterker M, Schneider D. Therapy of myasthenic crisis. Crit Care Med 1997;25(7):1228-1235.
- Moore KH, McKenzie PR, Kennedy CW, McCaughan BC. Thymoma: trends over time. Ann Thorac Surg 2001;72(1):203–207.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000;55(1):7-15.
- Sanders DB, El-Salem K, Massey JM, McConville J, Vincent A. Clinical aspects of MuSK antibody positive seronegative MG. Neurology 2003;60(12):1978-1980.
- Rosai J, Sobin L. Histological typing of tumours of the thymusIn: Rosai J, Sobin L, editors. World Health Organization, international histological classification of tumours. Berlin, New York: Springer; 1999. pp. 9-14.
- Masaoka A, Monden Y, Nakahara K, Tanioka T. Follow-up study of thymomas with special reference to their clinical stages. Cancer 1981;48(11):2485–2492.
- Utsumi T, Shiono H, Kadota Y, Matsumura A, Maeda H, Ohta M, Yoshioka Y, Koizumi M, Inoue T, Okumura M. Postoperative radiation therapy after complete resection of thymoma has little impact on survival. Cancer 2009;115(23):5413-20.
- Mangi AA, Wright CD, Allan JS, Wain JC, Donahue DM, Grillo HC, Mathisen DJ. Adjuvant radiation therapy for stage II thymoma. Ann Thorac Surg. 2002;74(4):1033-1037.
- Davidov-Lustig M, Klinghoffer (Strich) V, Kaplan-Dinur A, Steiner I. Memory abnormalities in myasthenia gravis: Possible fatigue of central nervous system cholinergic circuits. Autoimmunity 1992;14(1):85–86.
- Bartel PR, Lotz BP. Neuropsychological test performance and affect in myasthenia gravis. Acta Neurol Scand 1995;91(4):266–270.
- Hokkanen E, Toivakka E. Electroencephalographic findings in myasthenia gravis, 180 EEG recordings of 109 patients. Acta Neurol Scand 1969;45(5):556–567.
- Tartara A, Mola M, Manni R, Moglia A, Lombardi M, Poloni M, Piccolo G. EEG findings in 118 cases of myasthenia gravis. Rev EEG Neurolphysiol 1982;12(3):275–279.
- Kazis A, Pappa A, Xafenias D. EEG findings in myasthenia gravis. Electromyogr Clin Neurophysiol 1984;24(7):577–582.
- Fotiou F, Papakostopoulos D, Hamlatzis P. Changes in the pattern reversal visual evoked potential in myasthenia gravis. Electromyogr Clin Neurophysiol 1994;34(3):171–175.
- Kirby AW, Wiley RW, Harding TH. Cholinergic effects on the visual evoked potential. In: Cracco RQ, Bodis-Wollner I, editors, Evoked potentials, 1986: 296–305.
- Jech R, Ruzicka E. Brain stem auditory evoked potentials reflect central nervous system involvement in myasthenia gravis. J Neurol 1996;243(7):547–550.
- Lefvert AK, Pirskanen R. Acetylcholine-receptor antibodies in cerebrospinal fluid of patients with myasthenia gravis. Lancet 1977;2(8033):351–352.
- Adornato BT, Houff SA, Engel WK, Dalakas M, Madden DL, Sever JL. Abnormal immunoglobulin bands in cerebrospinal fluid in myasthenia gravis. Lancet 1978;2(8085):367–368.
- Keesey JC, Tourtellotte WW, Herrmann C, Andrews JM, Lindstrom J. Acetylcholine-receptor antibody in cerebrospinal fluid. Lancet 1978;1(8067):777.
- Kam-Hansen S. Asymptomatic oligoclonal CSF IgG and progressive increase of intrathecal IgG synthesis in a patient with myasthenia gravis treated with thymectomy – A 4-years follow-up. J Neurol Sci 1986;75(3):285–292.
- Quera-Salva MA, Guilleminault C, Chevret S, Troche G, Fromageot C, Crowe McCann C, Stoos R, de Lattre J, Raphael JC, Gajdos P. Breathing disorders during sleep in myasthenia gravis. Ann Neurol 1992;31(1):86–92.
- Eckert DJ, Jordan AS, Merchia P, Malhotra A. Central sleep apnea: pathophysiology and treatment. Chest 2007;131(2):595-607.
- Malhotra A, White DP. Obstructive sleep apnoea. Lancet 2002;360(9328):237-245.
- Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Rein J, Vela-Bueno A, Kales A. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med 2001 ;163(3 Pt 1):608-613.
- Bixler EO, Vgontzas AN, Ten Have T, Tyson K, Kales A. Effects of age on sleep apnea in men: I. Prevalence and severity. Am J Respir Crit Care Med 1998;157(1):144–148.
- Papazian O. Rapid eye movement sleep alterations in myasthenia gravis. Neurology 1976;26(4):311–316.
- Mennuni G, Morante MT, Di Meo L, Scoppetta C, Strusi L, Neri G, Laudisio A, Mazza S, Bergonzi P. Myasthenia and sleep. Schweiz Arch Neurol Neurochir Psychiatr 1983;133(2):193-203.
- Kryger M, Roth T, Dement W. Principles and practice of sleep medicine (3rd ed.). Philadelphia: Saunders. 2000.
- Salloum A, Rowley JA, Mateika JH, Chowdhuri S, Omran Q, Badr MS. Increased propensity for central apnea in patients with obstructive sleep apnea: effect of nasal continuous positive airway pressure. Am J Respir Crit Care Med 2010;181(2):1891-1893.
- Happe S, Klösch G, Zeitlhofer J. Perception of dreams and subjective sleep quality in patients with myasthenia gravis. Neuropsychobiology 2004;50(1):21-27.
- Bogousslavsky J, Regli F, Doret AM, Fulpius BW, Ostinelli B, Rabinowicz T, Ruzicka J. Encephalopathy, peripheral neuropathy, dysautonomia, myasthenia gravis, malignant thymoma, and antiacetylcholine receptor antibodies in the CSF. Eur Neurol 1983;22(5):301-306.
- Anderson NE, Hutchinson DO, Nicholson GJ, Aitcheson F, Nixon JM. Intestinal pseudo-obstruction, myasthenia gravis, and thymoma. Neurology 1996;47(4):985–987.
- Douchet MP, Quiring E, Bronner F, Vi-Fane R, Messier M, Chauvin M, Warter JM. Paradoxal lowering of parasympathetic indices in myasthenic patients. Arch Mal Coeur Vaiss 1999;92(6):711-717.
- Tan CK, Ng HS, Ho JS, Theobald DM, Lim YC. Acute intestinal pseudo-obstruction due to malignant thymoma. Singapore Med J 1993;34(2):175–178.
- Knezevic W, Bajada S. Peripheral autonomic surface potential. A quantitative technique for recording sympathetic conduction in man. J Neurol Sci 1985;67(2):239-251.
- Stoica E, Enulescu O. Deficiency of sympathetic nervous system function in myasthenia. J Auton Nerv Syst 1992;38(1):69-76.
- Fleisher LA, Beckman JA, Brown KA. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery: A Report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery. J Am Coll Cardiol 2007; 50(17):e159-e241
- Baraka A. Anesthesia and myasthenia gravis. Can J Anaesth 1992;39(5 Pt 1):476-486.
- Seifert HS, Smith DS. Neuroanesthesia and neurologic disease In: Longnecker DE, Murphy FL, Eds.; Introduction to Anesthesia. 9th ed. WB Saunders Co Philadelphia, PA: 1997, p. 400-414
- Dierdorf SF. Anesthesia for patients with rare and coexisting disease In: Barash PG, Cullen BF, Stoelting RK, Eds.; Clinical Anesthesia. 4th ed. Philadelphia, PA: 2001, p. 491-520
- Kunst G, Graf BM, Schreiner R. Differential effects of sevoflurane, isoflurane, and halothane on Ca2+ release from the sarcoplasmic reticulum of skeletal muscle. Anesthesiology 1999;91(1):179-186.
- Clark M, Brunick A- Nitrous Oxide Interaction with the Body, em: Clark M, Brunick A - Nitrous Oxide and Oxygen Sedation. St. Louis, Mosby, 1999;103-114.
- Saito Y, Sakura S, Takatori T, Kosaka Y. Epidural anesthesia in a patient with myasthenia gravis. Acta Anaesthesiol Scand 1993;37(5):513-515.
- Akpolat N, Tilgen H, Gürsoy F, Saydam S, Gürel A. Thoracic epidural anaesthesia and analgesia with bupivacaine for transsternal thymectomy for myasthenia gravis. Eur J Anaesthesiol, 1997;14(2):220-223.
- Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol 1997;53(2):199–237.
- Tsuneki H, Kimura I, Dezaki K, Kimura M, Sala C, Fumagalli G. Immunohistochemical localization of neuronal nicotinic receptor subtypes at the pre- and postjunctional sites in mouse diaphragm muscle. Neurosci Lett 1995;196(1-2):13–16.
- Tzartos SJ, Sophianos D, Efthimiadis A. Role of the main immunogenic region of acetylcholine receptor in myasthenia gravis. An Fab monoclonal antibody protects against antigenic modulation by human sera. J Immunol 1985;134(4):2343–2349.
- Sala C, Kimura I, Santoro G, Kimura M, Fumagalli G. Expression of two neuronal nicotinic receptor subunits in innervated and denervated adult rat muscle. Neurosci Lett 1996;215(2):71–74.
- Britto LR, Hamassaki-Britto DE, Ferro ES, Keyser KT, Karten HJ, Lindstrom JM. Neurons of the chick brain and retina expressing both alpha-bungarotoxin-sensitive and alpha-bungarotoxin-insensitive nicotinic acetylcholine receptors: an immunohistochemical analysis. Brain Res 1992;590(1–2):193–200.
- Barrantes GE, Rogers AT, Lindstrom J, Wonnacott S. Alpha- Bungarotoxin binding sites in rat hippocampal and cortical cultures: initial characterisation, colocalisation with alpha 7 subunits and up-regulation by chronic nicotine treatment. Brain Res 1995; 672(1– 2):228–236.
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci 2001;21(20):7993–8003.
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 1994;79(4):705–715.
- Lips K.S., Pfeil U., Kummer W. Coexpression of alpha 9 and alpha 10 nicotinic acetylcholine receptors in rat dorsal root ganglion neurons. Neuroscience 2002;115 (1):1–5.
- Drescher DG, Ramakrishnan NA, Drescher MJ, Chun W, Wang X, Myers SF, Green GE, Sadrazodi K, Karadaghy AA, Poopat N, Karpenko AN, Khan KM, Hatfield JS. Cloning and characterization of alpha9 subunits of the nicotinic acetylcholine receptor expressed by saccular hair cells of the rainbow trout (Oncorhynchus mykiss). Neuroscience 2004;127(3):737–752.
- Lukas RJ, Changeux JP, Le Novère N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 1999;51(2):397–401.
- Nelson ME, Wang F, Kuryatov A, Choi CH, Gerzanich V, Lindstrom J. Functional properties of human nicotinic AChRs expressed by IMR-32 neuroblastoma cells resemble those of alpha3beta4 AChRs expressed in permanently transfected HEK cells. J Gen Physiol 2001;118(5):563– 582.
- Colquhoun LM, Patrick JW. Pharmacology of neuronal nicotinic acetylcholine receptor subtypes. Adv Pharmacol 1997;39:191–220.
- Lennon VA, Lindstrom JM, Seybold ME. Experimental autoimmune myasthenia: a model of myasthenia gravis in rats and guinea pigs. J Exp Med 1975;141(6):1365–1375.
- Vernino S. Neuronal acetylcholine receptor autoimmunity. Ann N Y Acad Sci. 2008;1132:124-8.
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000;343(12):847–855.
- Goldstein DS, Holmes C, Dendi R, Li ST, Brentzel S, Vernino S. Pandysautonomia associated with impaired ganglionic neurotransmission and circulating antibody to the neuronal nicotinic receptor. Clin Auton Res 2002;12(4):281–285.
- Lennon VA, Ermilov LG, Szurszewski JH, Vernino S. Immunization with neuronal nicotinic acetylcholine receptor induces neurological autoimmune disease. J Clin Invest 2003;111(6):907–913.
- Nguyen VT, Ndoye A, Grando SA. Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am J Pathol 2000;157(4):1377–1391.
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001;411(6835):269–276.
- Lindstrom J, Anand R, Peng X, Gerzanich V, Wang F, Li Y. Neuronal nicotinic receptor subtypes. In: Diversity of Interacting Receptors 1995; 100–116.
- Tüzün E. Neuronal acetylcholine receptor α9-subunit: A possible central nervous system autoantigen. Medical Hypotheses 2006;67(3):561-565.
- Briani C, Doria A, Ruggero S, Toffanin E, Luca M, Albergoni MP, D’Odorico A, Grassivaro F, Lucchetta M, De Lazzari F, Balzani I, Battistin L, Vernino S. Antibodies to muscle and ganglionic acetylcholine receptors (AchR) in celiac disease. Autoimmunity 2008;41(1):100-104.
- Heilbronn E, Haggblad J, Kubat B. Antibodies to the nicotinic acetylcholine receptor, obtained from serum of myasthenic patients, may decrease acetylcholine release from rat hippocampal nerve endings in vitro. Ann NY Acad Sci 1981;377:198–207.
- Weidenfeld J, Bodoff M, Saphier D, Brenner T. Further studies on the stimulatory action of nicotine on adrenocortical function in the rat. Neuroendocrinology 1989;50(2):132–138.
- Wang Z, Low PA, Jordan J, Freeman R, Gibbons CH, Schroeder C, Sandroni P, Vernino S. Autoimmune autonomic ganglionopathy: IgG effects on ganglionic acetylcholine receptor current. Neurology 2007;68(22):1917-1921.
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol 1995;48(4):774–782.
- Picciotto MR, Caldarone BJ, Brunzell DH, Zachariou V, Stevens TR, King SL Neuronal nicotinic acetylcholine receptor subunit knockout mice: physiological and behavioral phenotypes and possible clinical implications. Pharmacol Ther 2001;92(2-3):89–108.
- Li M, Lester HA. Ion channel diseases of the central nervous system. CNS Drug Rev 2001;7(2):214–240.
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 2001;31(1):131–141.
- Barkai E, Hasselmo M. Acetylcholine and associative memory in the piriform cortex. Molecular Neurobiology 1997;15(1):17–29.
- Chitnis S, Rao J. Rivastigmine in Parkinson’s disease dementia. Expert Opin Drug Metab Toxicol 2009;5(8):941-955.
- Darreh-Shori T, Jelic V. Safety and tolerability of transdermal and oral rivastigmine in Alzheimer’s disease and Parkinson’s disease dementia. Expert Opin Drug Saf 2010;9(1):167-176.
- Shiono H, Roxanis I, Zhang W, Sims GP, Meager A, Jacobson LW, Liu JL, Matthews I, Wong YL, Bonifati M, Micklem K, Stott DI, Todd JA, Beeson D, Vincent A, Willcox N. Scenarios for autoimmunization of T and B cells in myasthenia gravis. Ann N Y Acad Sci 2003;998:237- 256.
- Roxanis I, Micklem K, McConville J, Newsom-Davis J, Willcox N. Thymic myoid cells and germinal center formation in myasthenia gravis; possible roles in pathogenesis. J Neuroimmunol 2002;125(1- 2):185-197.
- Delpy L, Douin-Echinard V, Garidou L, Bruand C, Saoudi A, Guéry JC. Delpy L, Douin-Echinard V, Garidou L, Bruand C, Saoudi A, Guéry JC. Estrogen enhances susceptibility to experimental autoimmune myasthenia gravis by promoting type 1-polarized immune responses. J Immunol 2005;175(8):5050–5057.
- Ostlie N, Milani M, Wang W, Okita D, Conti-Fine BM. Absence of IL-4 facilitates the development of chronic autoimmune myasthenia gravis in C57BL/6 mice. J Immunol 2003;170(1):604–612.
- Yarilin D, Duan R, Huang YM, Xiao BG. Dendritic cells exposed in vitro to TGF-beta1 ameliorate experimental autoimmune myasthenia gravis. Clin Exp Immunol 2002;127(2):214–219.
- Lechin F, van der Dijs B, Pardey-Maldonado B, John E, Jimenez V, Orozco B, Baez S, Lechin ME. Enhancement of noradrenergic neural transmission: an effective therapy of myasthenia gravis: a report on 52 consecutive patients. J Med 2000;31(5-6):333-361.
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve— an integrative interface between two supersystems: the brain and the immune system, Pharmacol Rev 2000;52(4):595–638.
- Bedoui S, Miyake S, Straub RH, von Hörsten S, Yamamura T. More sympathy for autoimmunity with neuropeptide Y?. Trends Immunol 2004;25(10):508–512.
- Kosec D, Vidi?-Dankovi? B, Isakovi? K, Leposavi? G. Alterations in the thymopoiesis in experimental autoimmune myasthenia gravis. Int J Neurosci 2005;115(4):461-477.
- Sun Y, Qiao J, Lu CZ, Zhao CB, Zhu XM, Xiao BG. Increase of circulating CD4+CD25+ T cells in myasthenia gravis patients with stability and thymectomy. Clin Immunol 2004;112(3):284–289.
- Liu R, La Cava A, Bai XF, Jee Y, Price M, Campagnolo DI, Christadoss P, Vollmer TL, Van Kaer L, Shi FD. Cooperation of invariant NKT cells and CD4+CD25+ T regulatory cells in the prevention of autoimmune myasthenia. J Immunol 2005;175(12):7898–7904.
- Lennon VA, Seybold ME, Lindstrom JM, Cochrane C, Ulevitch R. Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J Exp Med. 1978;147(4):973-983.
- Medof ME, Walter EI, Rutgers JL, Knowles DM, Nussenzweig V. Identification of the complement decay-accelerating factor (DAF) on epithelium and glandular cells and in body fluids. J Exp Med 1987;165(3):848–864.
- McNearney T, Ballard L, Seya T, Atkinson JP. Membrane cofactor protein of complement is present on human fibroblast, epithelial, and endothelial cells. J Clin Invest 1989;84(2):538–545.
- Meri S, Waldmann H, Lachmann PJ. Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Lab. Invest. 1991;65(5):532–537.
- Conti-Tronconi B, Tzartos S, Lindstrom J. Monoclonal antibodies as probes of acetylcholine receptor structure. 2. Binding to native receptor. Biochemistry 1981;20(8):2181–2191.
- Gomez CM, Richman DP. Anti-acetylcholine receptor antibodies directed against the alpha-bungarotoxin binding site induce a unique form of experimental myasthenia. Proc Natl Acad Sci USA 1983;80(13):4089–4093.
- Adikari SB, Lian H, Link H, Huang YM, Xiao BG. Interferon-gammamodified dendritic cells suppress B cell function and ameliorate the development of experimental autoimmune myasthenia gravis. Clin Exp Immunol 2004;138(2):230–236.
- Duan RS, Adikari SB, Huang YM, Link H, Xiao BG. Protective potential of experimental autoimmune myasthenia gravis in Lewis rats by IL-10-modified dendritic cells. Neurobiol Dis 2004;16(2):461– 467.
- Cohen-Kaminsky S, Jambou F. Prospects for a T-cell receptor vaccination against myasthenia gravis. Expert Rev Vaccines 2005;4(4):473–492.
- Dayan M, Sthoeger Z, Neiman A, Abarbanel J, Sela M, Mozes E. Immunomodulation by a dual altered peptide ligand of autoreactive responses to the acetylcholine receptor of peripheral blood lymphocytes of patients with myasthenia gravis. Hum Immunol 2004;65(6):571–577.
- Urbatsch IL, Sterz RK, Peper K, Trommer WE. Antigen-specific therapy of experimental myasthenia gravis with acetylcholine receptor-gelonin conjugates in vivo. Eur J Immunol 1993;23:776–779.
- Brenner T, Hamra-Amitay Y, Evron T, Boneva N, Seidman S, Soreq H. The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB J 2003;17(2):214–222.
- Soreq H, Seidman S. Acetylcholinesterase– new roles for an old actor. Nat. Rev. Neurosci 2001;2(4):294–302.
- Motomura M, Lang B, Johnson I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti P/Q and anti-N-type calcium channel autoantibodies in the Lambert–Eaton myasthenic syndrome. J Neurol Sci 1997;20(147):35–42.
- Tagami H, Imamura S, Noguchi S, Nishitani H. Coexistence of peculiar pemphigus, myasthenia gravis and malignant thymoma. Dermatologica 1976;152(3):181-190.
- Izumi Y, Kinoshita I, Kita Y, Toriyama F, Taniguchi H, Motomura M, Yoshimura T. Myasthenia gravis with diffuse alopecia areata and pemphigus foliaceus. J Neurol 2002;249(10):1455-1456.
- Lennon VA, Kryzer TJ, Griesmann GE, O’Suilleabhain PE, Windebank AJ, Woppmann A, Miljanich GP, Lambert EH. Calcium-channel antibodies in Lambert-Eaton myasthenic syndrome and other paraneoplastic syndromes. N Engl J Med 1995;332(22):1467–1474.
- Lambert EH, Lennon VA. Selected IgG rapidly induces Lambert– Eaton myasthenic syndrome in mice: complement independence and EMG abnormalities. Muscle Nerve 1988;11(11):1133–1145.
- O’Suilleabhain P, Low PA, Lennon VA. Autonomic dysfunction in the Lambert–Eaton myasthenic syndrome: serologic and clinical correlates. Neurology 1998;50 (1):88–93.
- Lorusso L, Hart IK, Ferrari D, Ngonga GK, Gasparetto C, Ricevuti G. Autonomic paraneoplastic neurological syndromes. Autoimmunity Reviews 2007;6(3):162-168.
- Shillito P, Molenaar PC, Vincent A, Leys K, Zheng W, van den Berg RJ, Plomp JJ, van Kempen GT, Chauplannaz G, Wintzen AR, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol 1995;38(5):714-722.
- Sciamanna MA, Griesmann GE, Williams CL, Lennon VA. Nicotinic acetylcholine receptors of muscle and neuronal (α7) types coexpressed in a small cell lung carcinoma. J Neurochem 1997;69(6):2302–2311.
- Griesmann GE, Harper CM, Lennon VA. Paraneoplastic myasthenia gravis and lung carcinoma: distinction from Lambert-Eaton syndrome and hypothesis of aberrant muscle acetylcholine receptor (AChR) expression. Muscle Nerve 1998;7(Suppl.):S122.
- Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci USA 1990;87(9):3294–3298.
- Codignola A, Tarroni P, Cattaneo MG, Vicentini LM, Clementi F, Sher E. Serotonin release and cell proliferation are under the control of α-bungarotoxin-sensitive nicotinic receptors in small-cell lung carcinoma cell lines. FEBS Lett 1994;342(3):286–290.
- Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R, Ramió-Torrentà L, Graus F. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008;131(Pt 10):2553-2563.
- Camdessanché JP, Antoine JC, Honnorat J, Vial C, Petiot P, Convers P, Michel D. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain 2002;125(Pt 1):166–175.
- Rosenblum MK. Paraneoplasia and autoimmunologic injury of the nervous system: the anti-Hu syndrome. Brain Pathol 1993;3(3):199– 212.
- Okumura M, Fujii Y, Shiono H, Inoue M, Minami M, Utsumi T, Kadota Y, Sawa Y. Immunological function of thymoma and pathogenesis of paraneoplastic myasthenia gravis. Gen Thorac Cardiovasc Surg 2008;56(4):143-150.
- Yu Z, Kryzer TJ, Griesmann GE, Kim K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol 2001;49(2):146–154.
- Antoine JC, Honnorat J, Camdessanché JP, Magistris M, Absi L, Mosnier JF, Petiot P, Kopp N, Michel D. Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol 2001;49(2):214–221.
- Darnell RB. Paraneoplastic neurological disorders: windows into neuronal function and tumor immunity. Arch Neurol 2004;61(1):30– 32.
- Becher B, Prat A, Antel JP. Brain–immune connection: immunoregulatory properties of CNS-resident cells. Glia 2000;29(4):293–304.
- Fattorossi A, Battaglia A, Buzzonetti A, Minicuci G, Riso R, Peri L, Scambia G, Evoli A. Thymopoiesis, regulatory T cells, and TCRVbeta expression in thymoma with and without myasthenia gravis, and modulatory effects of steroid therapy. J Clin Immunol 2008;28(2):194-206.
- Low PA, Suarez GA. Diabetic neuropathies. Baillieres Clin Neurol 1995;4(3):401-425.
- Manfredi R, Fasulo G, Fulgaro C, Sabbatani S. Associated thyreoiditis, myasthenia gravis, thymectomy, Chron’s disease, and erythema nodosum: pathogenetic and clinical correlations, immune system involvement, and systemic infectious complications. Rheumatol Int 2008;28(11):1173-1175.
- Kubota A, Komiyama A, Tanigawa A, Hasegawa O. Frequency and clinical correlates of vitiligo in myasthenia gravis. J Neurol 1997;244(6):388-389.
- Kai Y, Ohyagi Y, Inoue I, Higashino T, Yamada T, Kira J. A patient with limb-girdle type myasthenia gravis and atopic dermatitis, both of which improved after thymectomy. Rinsho Shinkeigaku 2000;40(4):405-408.
- Angelucci E, Cesarini M, Vernia P. Successful resolution of pneumonia developed in a patient affected by Crohn’s disease, rheumatoid arthritis, myasthenia gravis and recurrent uveitis during concomitant treatment with tumour necrosis factor alpha inhibitors and conventional immunosuppressive drugs. Rheumatol Int 2010;30(7):977-978.
- Tola MR, Casetta I, Granieri E, Caniatti LM, Monetti VC, Pascarella R. Systemic lupus erythematosus related recurrent transverse myelitis in a patient with myasthenia gravis and multiple sclerosis. Eur Neurol 1996;36(5):327-328.
- El Sayed F, Dhaybi R, Ammoury A, Chababi M, Bazex J. Myasthenia gravis with cutaneous polyarteritis nodosa. Clin Exp Dermatol 2006;31(2):215-217.
- Mineo TC, Biancari F, D’Andrea V. Myasthenia gravis, psychiatric disturbances, idiopathic thrombocytopenic purpura, and lichen planus associated with cervical thymoma. J Thorac Cardiovasc Surg 1996;111(2):486-487.
- Szurszewski JH, Miller SM. Physiology of prevertebral ganglia. In Physiology of the gastrointestinal tract. 3rd edition. Johnson LR. editor. Raven Press. New York, New York, USA 1994, 795–877.
- Paradis CM, Friedman S, Lazar RM, Kula RW. Anxiety disorders in a neuromuscular clinic. Am J Psychiatry 1993;150(7):1102–1104.
- Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep 1999;22(5):667-689.

