Keywords
Hepatic fibrosis, Caroli's disease
Case Description
Both cases were presented to-National Liver Institute, Menoufia University, Egypt- with one year interval from September 2015 to August 2016.
Case 1
A 22 years old gentleman had presented to National Liver Institute hospital with recurrent attacks of mild upper gastrointestinal bleeding during the last month. He was completely free before the bleeding events and has relevant family history with no consanguinity. The patient was vitally stable, mild pallor, hepatosplenomegaly, and ascites were clinically detected. Upper gastrointestinal endoscopy with band ligation of grade 3 esophageal varices was the intervening maneuver of choice. Laboratory investigations showed negative results for hepatitis B surface antigen (HBsAg) anti-hepatitis B core (HBc)-IgG, anti-HBc-IgM, anti- EBV-IgM, anti-CMV-IgM, anti-HIV-Ab, and anti-HSV-IgM), and polymerase chain reaction (PCR) for HCV-RNA.
Serum ferritin and urinary copper were within normal ranges with normal slit lamp examination. Antinuclear antibody (ANA), anti-smooth muscle antibody (ASMA) antimitochondrial antibody (AMA) perinuclear anti-neutrophil cytoplasmic antigen (p-ANCA) and anti-liver-kidney microsomal antibody (anti-LKM-1) were negative. Abdominal ultrasound examination revealed: liver cirrhosis, splenomegaly and mild ascites, along with normal morphology of both kidneys. Doppler studies ruled out portal vein thrombosis, with portal vein diameter (13 mm), and reduced portal flow (11 m/ sec.). However, transient elastography had negated the presence of cirrhosis (F3). Ascitic fluid examination of serum ascites albumin (SAAG) ratio showed its portal hypertensive nature. Consequently, liver biopsy was done and revealing periportal and portoportal islands of fibrosis separating the parenchyma into nodules of variable sizes and containing bizarre shaped serpingeous pattern of bile ductules with no inflammatory cells (Figures 1 and 2). A picture was highly suggestive of latent form of congenital hepatic fibrosis. Another session of variceal band ligation was done and propranolol 20 mg three times a day was prescribed for the patient before discharge (Table 1).
| |
On admission |
One week later |
| Total bilirubin (mg/dl) |
1.1 |
1.1 |
| Direct bilirubin (mg/dl) |
0.7 |
0.6 |
| AST (IU/dl) |
25 |
22 |
| ALT (IU/dl) |
29 |
21 |
| ALP (IU/dl) |
70 |
60 |
| GGT (IU/dl) |
50 |
66 |
| Total protein (gm/dl) |
6.8 |
5.1 |
| Albumin (gm/dl) |
3.9 |
4.1 |
| Prothrombin concentration |
90% |
90.8% |
| Hb (g/dl) |
8 |
10.1 |
| WBC (103/dl) |
4.1 |
4.0 |
| Platelets (103/dl) |
90 |
120 |
AST: Aspartate Transaminase, ALT: Alanine Transaminase, ALP: Alkaline Phosphatase, GGT: Gamma Glutamyl Transferase, HB: Haemoglobin, WBCs: White Blood Cells
Table 1 Laboratory data of case 1.
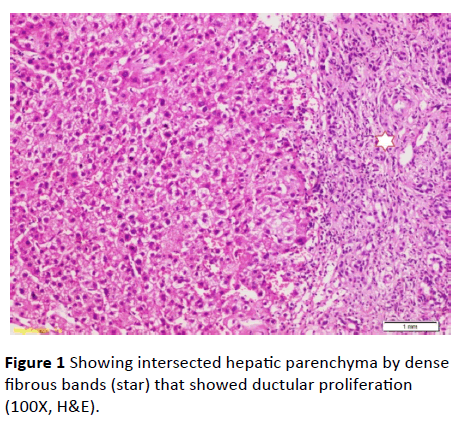
Figure 1 Showing intersected hepatic parenchyma by dense fibrous bands (star) that showed ductular proliferation (100X, H&E).
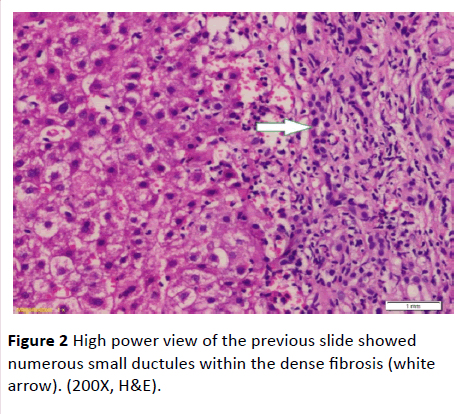
Figure 2 High power view of the previous slide showed numerous small ductules within the dense fibrosis (white arrow). (200X, H&E).
Case 2
A 19-year-old lady had presented to National Liver Institute hospital with recent jaundice and abdominal distention for the previous three month. The patient reported a history of two similar attacks in the last two years. She had no history suggestive of exposure to hepatotoxic substances. No family history of liver or kidney disease and sanguinity is absent. The patient was mildly feverish (38.5) and having jaundice palpable liver and spleen along with mild ascites. Ascitic fluid examination was sterile and demonstrated high serum to ascites albumin (SAAG) ratio. The fever rapidly disappeared on antibiotic treatment that was immediately initiated in the form of Cefotaxim 1 g IV daily. Laboratory investigations showed hyperbilirubinemia, with elevated cholestatic enzymes and normal kidney functions (Table 2). Also, hepatitis B surface antigen (HBsAg) anti-hepatitis B core (HBc)-IgG, anti-HBc-IgM, anti- EBV-IgM, anti-CMV-IgM, anti-HIV-Ab, and anti-HSV-IgM), and polymerase chain reaction (PCR) for HCV-RNA, all were negative. Serum ferritin and urinary copper were within normal ranges with normal slit lamp examination. Antinuclear antibody (ANA), anti-smooth muscle antibody (ASMA) antimitochondrial antibody (AMA) perinuclear anti-neutrophil cytoplasmic antigen (p-ANCA) and anti-liver-kidney microsomal antibody (anti-LKM-1) were negative. Abdominal ultrasound examination demonstrated absence of echogenic hepatomegaly, splenomegaly and mild ascites, with normally appearing both kidneys. Doppler studies showed reduced portal flow (12 cm/sec), no thrombosis and dilated portal vein (15 mm). Values in Transient elastography were comparable to F3. Liver biopsy was done, revealing the presence of disfigured architecture with numerous thick fibrous septa. The fibrous septa contain numerous abnormally shaped bile ducts with bile cholangiolar proliferation and evidently periductular polymorph infiltration. Interface was mainly made by bile ductular proliferation (ductular interface). Parenchyma was formed of normal looking hepatocytes except for occasional canalicular cholestasis (Figures 3 and 4). The histological picture was consistent with ductal plate malformation with ascending cholangitis and no true cirrhosis. Furtherly, Orcien staining had documented the absence of biliary cirrhosis. The patient reported no history of bleeding and refused to have an upper endoscopy.
| |
On admission |
One week later |
| Total Bilirubin (mg/dl) |
3.9 |
2.0 |
| Direct Bilirubin (mg/dl) |
1.9 |
0.9 |
| Albumin (gm/dl) |
3.8 |
3.9 |
| AST (IU/l) |
39 |
25 |
| ALT (IU/l) |
41 |
20 |
| AP (IU/l) |
433 |
441 |
| GGT (IU/l) |
106 |
101 |
| Hb (gm/dl) |
10.3 |
9.5 |
| WBC (103/dl) |
6.8 |
8.7 |
| Platelets (103/dl) |
89 |
106 |
| Blood Urea (mg/dl) |
13 |
20 |
| S. Creatinine (mg/dl) |
0.6 |
0.8 |
| INR % |
1.6 |
1.2 |
AST: Aspartate Transaminase, ALT: Alanine Transaminase, ALP: Alkaline Phosphatase, GGT: Gamma Glutamyl Transferase, HB: Haemoglobin, WBCs: White Blood Cells, INR: International Normalized Ratio
Table 2 Laboratory data of case 2.
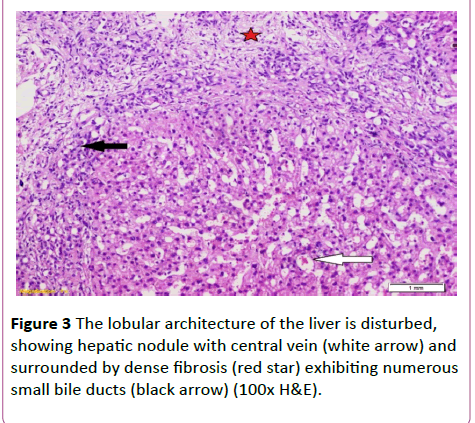
Figure 3 The lobular architecture of the liver is disturbed, showing hepatic nodule with central vein (white arrow) and surrounded by dense fibrosis (red star) exhibiting numerous small bile ducts (black arrow) (100x H&E).
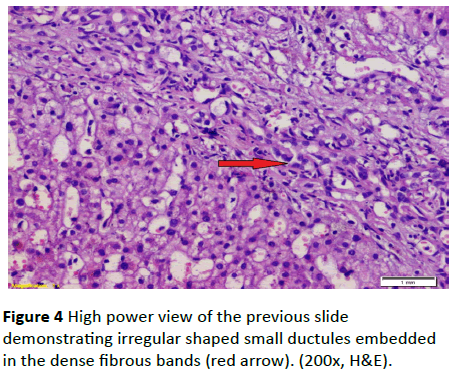
Figure 4 High power view of the previous slide demonstrating irregular shaped small ductules embedded in the dense fibrous bands (red arrow). (200x, H&E).
Discussion
Morphogenesis of the biliary tree begins as early as week 9 to 10 of gestation when precursor hepatocytes encasing the mesenchyme of the portal vein became compressed and united through various cytokeratins to form one layer then two layers of cells [1]. In response to unknown signaling, biliary development progresses by formation of a cleft throughout the two layers of cells known as the ductal plate. This process extends proximally from the porta hepatis and parallels the portal vein branches in the liver parenchyma. Afterwards, these ductal plates undergo morphological remodeling; thus, creating the intrahepatic biliary ducts (IHBDs) [2]. Dysmorphogenesis of the ductal plates or ductal plate malformations (DPMs) can affect the segmental, septal, interlobular and the smaller intrahepatic branches of the biliary tree [3]. Congenital diseases of IHBDs are the correspondents of arrested remodeling of the ductal plates at different levels of the biliary tree [4].
CHF is named by Kerr and since that time many cases had been reported, but the true incidence of CHF is unknown [5]. A recent report mentioned an incidence of 1 in 10.000 to 20.000 live births [6]. It depicts dysmorphogenesis of the smallest immature intrahepatic bile ducts. The lack of remodeling of these immature IHBDs by a non-specific necroinflammatory process may undergo a progressive destruction and disappearance [7]. This progressive destructive cholangiopathy had shown to be coupled by progressive periportal and perilobular fibrosis [8]. Uniquely, this fibrosis is a non-resolving process that might be explained by the abundant connective tissue growth factor evenly retained in heparin sulfate proteoglycan in the fibrous septa [9]. In addition, the rate of both bile duct damage and fibrosis formation has shown to be variable and time dependant [10]. Therefore, hepatic fibrosis and portal hypertension are progressing with age. Faulty remodeling of the larger intrahepatic biliary ducts depicts another closely related fibrocystic variant; Caroli's disease. CHF can be combined with dysmorphic larger intrahepatic bile ducts and symbolized as the Caroli's syndrome to differentiate it from Caroli’s disease [11].
In the most, CHF is coupled with similar fibrocystic renal affection, clinically expressed as inheritable polycystic kidney diseases. Autosomal recessive polycystic kidney disease (ARPKD) is the classic kidney abnormality in conjunction with CHF; the autosomal dominant variety had also been described in older patients [12]. Many other pulmonary, cardiac, ocular and brain malformations had been rarely mentioned in association with ductal plate dysmorphogenesis [10]; all could be excluded in the cases presented in this report by clinical examination and investigations.
ARPKD is invariably coupled with histopathological features relevant to CHF. However, clinical expression of CHF is independently correlated with the severity of kidney affection. Recent researches suggested that the severe kidney/severe hepato-biliary disease phenotype may constitute up to 40% of the ARPKD patients [13]. In a recent systematic review that included a total of 1230 patients with CHF; only 9.5% of cases were isolated CHF while the majority were ARPKD/CHF [14]. In this report, the two patients were having no other relevant disease process and considered as having isolated CHF.
While autosomal recessive inheritance is the committed feature of CHF, familial as well as sporadic cases had been proportionately reported [15]. Both cases reported here are probably examples of sporadic cases. Subclinical or milder forms might exist in other family members.
Dedicated research work revealed that ARPKD/CHF syndrome is caused by mutations in the PKHD1 gene. More than 750 mutations of the PKHD1 gene have been reported [16]. Fibrocystin is a large integral membrane protein encoded by the PKHD1 gene. Fibrocystin, also referred to as polyductin, is found in the epithelial cells of the renal and hepatic bile ducts [17]. Recently, deficient fibrocystin has been proposed to be linked to the pathogenesis of ARPCKD/CHF and about 45% of the PKHD1 mutations are anticipating to the lacking of fibrocystin [10].
Most of cases of CHF are diagnosed during childhood and adolescence. However, the disease can present itself as early as in neonates; also, some cases are reported in the fifth and sixth decades [18]. The age of presentation of ARPKD/CHF is similarly variable depending upon the degree of pathological affection of the liver and kidney [19]. In fact, the prognosis of ARPKD/CHF had been linked to the age of clinical expression of the disease; renal affection and prognosis are worst in the perinatal cases with one month mortality rate to 6 month mortality rate in the neonatal cases. Renal disease and prognosis are less severe in infantile and juvenile presentations [20]. Those with isolated hepatic disease are expectedly having better prognosis with exception of the cholangiotic variety prone to repeated biliary sepsis [21]. Both patients in this report had no clinical or radiological evidence of renal abnormalities, a fact that explains their presentation at an otherwise older age.
Basically, clinical presentations of CHF are the outcomes of portal hypertension with variceal bleeding of varying severity, splenomegaly, and thrombocytopenia [19]. Hepatomegaly with firm to hard liver on palpation, particularly the left lobe and nephromegaly in those having ARPKD are the additional physical findings in CHF [12]. Cholangitis is reported to be infrequent and can predominate the clinical picture even in absence of significant cystic biliary changes [22]. In the second case, repeated cholangitis suggested possibility of an element of Caroli’s syndrome versus cholangitic subtype of CHF. In this case; histopathological examination of liver tissue and Orcien staining had ruled out biliary cirrhosis. It’s noteworthy to describe clinical presentations of CHF as portal hypertensive predominance. Cholangitic predominance with cholestasis and recurrent cholangitis, mixed presentation and latency form that appears at a late age with hard hepatomegaly [22].
Unlike most chronic liver diseases, CHF is characteristically accompanied by preserved hepatocellular functions and features of progressive hepatic insufficiency are infrequently reported [10]. Development of ascites in the setting of CHF should prompt further searching for a secondary condition as portal vein thrombosis or dialysis related ascites [14]. The two patients in this report were having ascites with high SAG and absent secondary causes of ascites, thus representing the uncommon variant of CHF. An explanation for ascites particularly in older patients who are having extensive periportal and hepatic fibrosis is the increased resistance to portal blood flow that will be transmitted to the pre-portal venous system, forcing fluid out of these vessels to the peritoneal cavity and forming ascites.
Histopathological examination of liver tissue is the definitive prerequisite for diagnosing CHF [22]. Liver tissue shows fibrous septa periportally and interportally, the later causes false distortion of the lobular architecture with creation of different size nodules; a picture mimicking cirrhosis. Indeed, absence of porto-central vein bridging, absent regenerative nodules and normally looking hepatocytes would distinguish CHF from cirrhosis [23]. The fibrous septa contain minimal infiltration with inflammatory cells and characteristically large number of embryonic bile ducts attaining different configurations [23]. Among these configurations, cystic bile ducts, acting as a part of Caroli’s syndrome or being a cholangitic subtype of CHF, could be identified [22]. Ductal plate malformation is perceived as a portobiliary anomaly and portal vein could undergo abnormal branching, hypoplastic changes, also, cavernous transformation of the portal vein had been reported in association with CHF [13]. Limitations of liver tissue examination in CHF include the one lobe affection' variant, additionally, the divergent histological pattern [10].
Treatment for CHF is largely directed toward treating complications associated with portal hypertension and biliary infections; if present. The potential future medical therapies for fibrocystic disorders include Octreotide; a somatostatin analogue, Pioglitazone; a peroxisome proliferator activator receptor gamma agonist and Gefitinib, a tyrosine kinase have sh own effectiveness for both liver and kidney disease in animal models [24].
Up-to-date, there is no consensus on the indications of dual liver and kidney transplantation for APKD/CHF [25,26].
In more than one report, biliary sepsis contributes to significant number of deaths in ARPKD pediatric renal transplant recipients [14]. Therefore, some authors suggested combined liver-kidney transplantation aiming to improve the dominant mortality in ARPKD patients with ESRD and clinically significant CHF [14]. For cases with significant CHF, portosystemic shunting prior to isolated kidney transplantation could be an option [14], but data in the literature concerning this point are still controversial. A recent report from the University Medical Center Hamburg- Eppendorf; a major referral center for patients with combined kidney and liver transplantation including ARPKD, recommended dual liver and kidney transplantation in those having chronic kidney disease stage IV/ESRD and grade IV hepatic fibrosis with portal hypertensive varices [15]. Also, Portosystemic shunting was recommended as an option in those having advanced hepatic fibrosis and variceal bleeding if they have normal kidney function [16].
Conclusion
The age of presentation of CHF typically ranges between 1.8-14 years, both cases in this report were presented at a relatively older age. CHF can present as an isolated condition or can occur in the context of ARPKD or Caroli’s syndrome, also, can be inherited and can be a sporadic affection. Cases in this report are having isolated CHF and are possibly sporadic cases of the disease. Atypically, they have high SAG ascites along with patent portal veins and no cirrhotic changes on liver biopsy. The first case has portal hypertensive predominance while the second case is portraying a mild cholangitic/portal hypertensive variant.
Author Contributions
All authors listed in this manuscript contributed significantly to the development and writing of this manuscript.
Acknowledgment
We are deeply indebted to all our colleagues in Hepatology, Clinical Pathology, Pathology, and Radiology departments in National Liver Institute hospital, Menofia University, Egypt for their tremendous efforts affording the best management to the reported cases.
18492
References
- Coffinier C, Gresh L, Fiette L (2002) System morphogenesis defects and liver dysfunction upon targeted deletion of HNF1β. Development 129: 1829-1883.
- Sparks E, Huppert K, Brown M (2010) Notch signaling regulate formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 51: 1391-1400.
- Raynaud P, Tate J, Callens C (2011) A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology 53: 1959-1966.
- Desmet VJ (1992) Congenital diseases of intrahepatic bile ducts: Variations on the theme "ductal plate malformation". Hepatology 16: 1069-1083.
- Kerr DN, Harrison CV, Sherlock S (1961) Congenital hepatic fibrosis. Quart jmed 30: 91-117.
- Dhameja N, Rai V, Singh R (2016) Congenital hepatic fibrosis : Report on two cases and its clinicopathological correlation.AnnPatholLab Med 3: 90-94.
- Veigel MC, Prescott-Focht J, Rodriguez MG (2009) Fibropolycystic liver disease in children. Pediatr Radiol 39: 317-327.
- Nakanuma Y (2012) Tutorial review for understanding of cholangiopathy. Int J Hepatol 9.
- Ozaki S, Sato Y, Yasoshima M (2005) Diffuse expression of heparin sulfate proteoglycan and connective tissue growth factor in fibrous septa with many mast cells relate to unresolving hepatic fibrosis of congenital hepatic fibrosis. Liver Int 25: 817-828.
- Gunay-Aygun M, Tuchman M, Font-Montgomery E (2010) PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab 99: 160-173.
- Shenoy P1, Zaki SA, Shanbag P (2014) Caroli's syndrome with autosomal recessive polycystic kidney disease. Saudi J Kidney Dis Transpl 25: 840-843.
- Turkbey B, Ocak I, Daryanani K (2009) Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). PediatrRadiol 39: 100-111.
- Bayraktar Y (2011) Portal ductopathy: clinical importance and nomenclature. World J Gastroenterol 17: 1410-1415.
- Telega G, Cronin D, Avner E (2013) New approaches to the ARPKD patient with dual kidney-liver complications.Pediatr Transplant 17: 328-335.
- Srinath A, Shneider B (2012) Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease. J PediatrGastroenterolNutr 54: 580-587.
- Patel JN, Gupta S, Fauzdar M (2015) Congenital hepatic fibrosis associated with polycystic kidney disease.J Liver 4: 171.
- Bergmann C, Senderek J, Sedlacek B (2003) Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am SocNephrol 14: 76-89.
- Kim I, Fu Y, Hui H (2008) Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am SocNephrol 19: 455-468.
- Wen G (2011) Congenital hepatic fibrosis in autosomal recessive polycystic kidney disease. Clin Trans Sci 4: 460-465.
- Shorbagi A, Bayraktar Y (2010) Experience of a single center with congenital hepatic fibrosis: A review of the literature. World J Gastroenterol 16: 683-690.
- Nazer D, Elzouki NH (2012) Congenital hepatic fibrosis. Textbook of clinical pediatrics. 2ndedn. Berlin-Heidelberg: Springer-Verlag 3: 2013-2016.
- Giouleme O, Nikolaidis N, Tziomalos K (2006) Ductal plate malformation and congenital hepatic fibrosis: Clinical and histological findings in four patients. Hepatol Res 2: 147-150.
- Jiang L, Fang P, Weemhoff J (2016) Evidence for a “Pathogenic Triumvirate” in congenital hepatic fibrosis in autosomal recessive polycystic kidney disease. Biomed Res Int.
- Sato Y, Ren XS, Nakanuma Y (2012) Caroli's disease: Current knowledge of its biliary pathogenesis obtained from an orthologous rat model. Int J Hepatol.
- Parkash A, Cheema H, Malik H (2016) Congenital hepatic fibrosis: Clinical presentation, laboratory features and management at a tertiary care hospital of Lahore JPMA 66: 984.
- Geramizadeh B, Keramati P, Bahador A (2010) Congenital hepatic fibrosis and need for liver transplantation. Int J Organ Transplant Med 1: 98-100.