Keywords
MELAS, mitochondrial encephalopathy, genetic diagnosis
Introducción
El síndrome MELAS (miopatía mitocondrial, encefalopatía, acidosis láctica y “stroke-like” episodios) es un síndrome descrito por primera vez por Pavlakis en 1984 y relacionado con una mutación en el ADN mitocondrial. Desde nuestro punto de vista el síndrome MELAS es aún poco conocido y dada la inexistencia de recomendaciones diagnósticas consensuadas se comenten errores en el diagnóstico con frecuencia. En este artículo se presenta un caso clínico que invita a realizar varias consideraciones al respecto.
Caso Clínico
Un varón diestro de 38 años entre cuyos antecedentes destacan dificultad auditiva e historia familiar de sordera y diabetes mellitus, debuta con fiebre, cefalea, confusión mental y crisis parciales con generalización secundaria. A continuación se hace evidente una focalidad persistente parieto-temporal derecha consistente en hemiparesia izquierda con hipoestesia asociada, hemianopsia homónima izquierda y desorientación espacial. Ante la sospecha de encefalitis herpética se inicia tratamiento antiviral con Aciclovir a dosis standard.
La tomografía computerizada de cráneo mostró calcificación bilateral de los ganglios basales y una hipodensidad en los lóbulos parietotemporal derechos (Figuras 1 y 2).
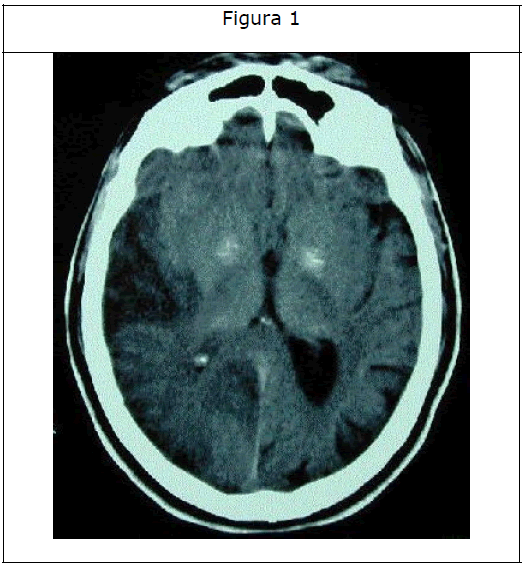
Figura 1
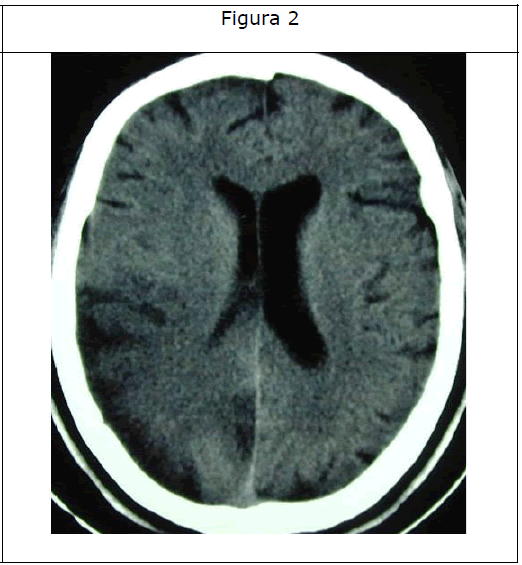
Figura 2
El líquido cefalorraquídeo analizado al tercer día de ingreso fue normal. La resonancia magnética craneal mostró un área de hiperintensidad parieto-témporo-occipital derecha córtico subcortical que no se corresponde con un territorio de irrigación vascular y atrofia difusa de la corteza cerebelosa (Figuras 3 y 4).
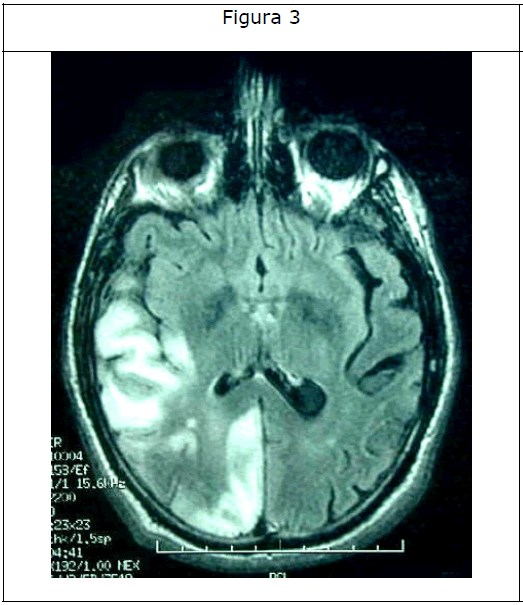
Figura 3
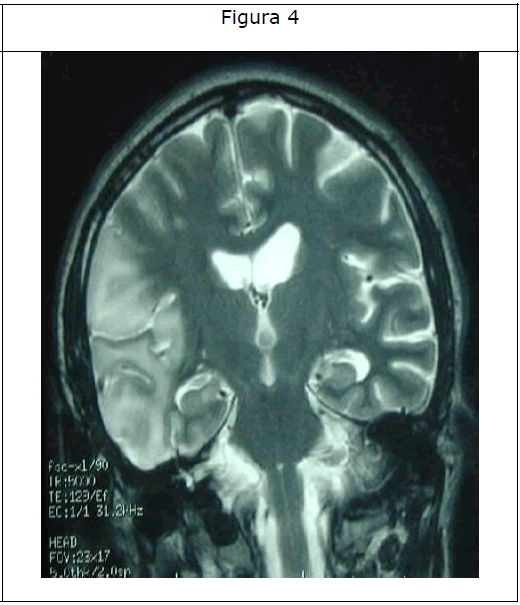
Figura 4
Un electroencefalograma ictal mostró ondas rítmicas de gran amplitud en la región parieto temporal derecha con un fondo de ondas lentas. El ácido láctico en la sangre y líquido cefalorraquídeo estaba dentro de los límites normales. Se practicó una biopsia muscular cuyo estudio neuropatológico mostró ausencia de tinción tricrómica y captación de la Citocromo-COxidasa (COX). El análisis del ADN mitocondrial detectó una mutación A3243G. Se amplió el estudio genético al resto de la familia (2 hermanas, 1 hermano y la madre), y la mutación se detectó en todos ellos pero en una proporción sanguínea muy variable, siendo incluso informada inicialmente la madre como negativa.
Discusión
Los pacientes portadores de la mutación suelen instaurar la clínica antes de los 40 años de edad. Hipoacusia, diabetes mellitus y esterilidad son los antecedentes personales y familiares más comunes. Crisis epilépticas, episodios tipo ictus (“stroke-like”), movimientos extrapiramidales, disfunción autonómica, cefalea, fiebre y confusión son posibles síntomas de la enfermedad. Dado que frecuentemente debutan con cefalea, confusión mental y fiebre no es infrecuente interpretar inicialmente el cuadro como una posible encefalitis herpética [1]; que en todo caso se ha de descartar mediante el análisis del líquido cefalorraquídeo.
Como explicación a los episodios “stroke-like” del MELAS se ha propuesto que pueden reflejar fenómenos de hiperexcitabilidad neuronal que incrementa la demanda energética y crear un disbalance energético entre los requerimientos y la disponibilidad de adenosina trifosfato debido a defectos de la fosforilación en poblaciones neuronales particularmente susceptibles causando necrosis cortical [2]. Sin embargo la naturaleza episódica de estos episodios persiste sin explicación.
Los niveles plasmáticos y licuorales de ácido láctico se encuentran típicamente elevados, pero en ocasiones, como en el caso presentado, pueden ser normales; por lo que no sirven para descartar un trastorno mitocondrial [3].
Una característica encefalográfica común a todas las encefalomiopatías mitocondriales es una actividad de fondo enlentecida [4]. Respuestas fotoparoxísticas y descargas periódicas lateralizadas también se pueden observar hasta varias semanas tras la instauración del brote [5]. Tanto la resonancia como la tomografía craneal suelen mostrar lesiones en áreas que típicamente no se corresponden con un territorio vascular. Las secuencias FLAIR (fluid attenuated inversion recovery) y difusión suelen mostrar imágenes hiperintensas multifocales córtico-subcorticales localizadas en los lóbulos témporo-occipitales que progresivamente se extienden hacia áreas corticales adyacentes más posteriores en las semanas siguientes a la instauración de los síntomas [6]. El hallazgo más común en las fases subagudas y crónicas son señales hiperintensas corticales en secuencia T1 compatibles con necrosis laminar cortical. [5]
Igualmente, estudios mediante HMPAO (HexaMethylPropyleneAmineOxime) SPECT seriados durante la evolución de un episodio “stroke-like”, los primeros mostraron hiperperfusión focal cuyo núcleo se situaba en la corteza correspondiente a la lesión radiológica para desplazarse en sucesivos estudios a regiones más posteriores [5].
Los estudios realizados mediante PET para conocer el metabolismo cerebral de pacientes con encéfalo-miopatías mitocondriales han demostrado que la recaptación cerebral de glucosa se encuentra descendida en todos ellos, especialmente en los lóbulos temporal y occipital, tanto si existe sintomatología neurológica como si no [7].
La biopsia muscular generalmente muestra muchas fibras “rojo-rasgadas”, la mayor parte de las cuales son positivas para COX [8]; sin embargo, la ausencia de hallazgos patológicos en la biopsia muscular tampoco puede descartar un síndrome MELAS dado que no en todos los casos la encefalopatía se acompaña de miopatía [9].
Las características de la genética mitocondrial son herencia materna, poliplasmia, heteroplasmia, segregación mitótica y efecto umbral que explican las peculiaridades hereditarias de las enfermedades provocadas por mutaciones en el ADN mitocondrial [10]. Varias mutaciones han sido descritas para el Síndrome MELAS, y éste podría ser uno de los factores que modulase las diferentes características clínicas y el pronóstico de cada familia. [11]. La mutación más común es la debida a la transición de A por G en la posición 3243 del ADN mitocondrial, que afecta a aproximadamente al 80% de los casos y se asocia con deficiencias en el complejo de la cadena respiratoria. Las razones por las cuales existe heterogeneidad fenotípica entre las diferentes familias con esta mutación aún no han sido clarificadas por completo aunque podrían relacionarse con la heteroplasmia y las diferentes proporciones mutacionales existentes en los diferentes tejidos. De este modo, pacientes en los que se analizan tanto muestras sanguíneas como musculares el porcentaje mutacional suele ser significativamente más bajo en la sangre llegando incluso a resultar indetectable por los métodos habituales, mientras que en músculo la mutación es claramente positiva [12].
Así, si nos encontramos con un caso como el presentado, en el que al estudiar una muestra sanguínea perteneciente a la madre del caso índice la mutación no fuera hallada, habría que plantearse si nos encontramos ante una mutación “de novo” o más probablemente que la mutación en las células sanguíneas de la madre ha sufrido un fenómeno de aclaramiento y presenta un porcentaje muy bajo.
1413
References
- De Toledo M, Diaz-Guzman J, Perez-Martinez DA, et al. MELAS syndrome masquerading as herpes encephalitis: genetic diagnosis. Rev Neurol 2001 Jul;16-31;33(2):148-50.
- Iizuka T, Sakai F, Suzuki N, Hata T, et al. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology 2002 Sep 24;59(6):816-24
- Ciafaloni E, Ricci E, Shanske S, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol 1992 Apr;31(4):391-8.
- Tulinius MH, Hagne I. EEG findings in children and adolescents with mitochondrial encephalomyopathies: a study of 25 cases. Brain Dev 1991 May;13(3):167-73.
- Iizuka, Takahiro MD; Sakai, Fumihiko MD; Kan, Shinichi MD; Suzuki, Norihiro MD. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology, Volume 61(9) 11 November 2003 pp 1238-1244
- Oppenheim C, Galanaud D, Samson Y, et al. Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J Neurol Neurosurg Psychiatry 2000 Aug;69(2):248-50.
- Molnar MJ, Valikovics A, Molnar S, et al. Cerebral blood flow and glucose metabolism in mitochondrial disorders. Neurology 2000 Aug 22;55(4):544-8.
- Tanahashi C, Nakayama A, Yoshida M, et al. MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol (Berl) 2000 Jan;99(1):31-8.
- Ozawa M, Nonaka I, Goto Y. Single muscle fiber analysis in patients with 3243 mutation in mitochondrial DNA: comparison with the phenotype and the proportion of mutant genome. J Neurol Sci.1998 Aug 14;159(2):170-5.
- Martin M, Campos Y, de Bustos F, et al. Genética molecular de las alteraciones de la cadena respiratoria mitocondrial. Rev. Neurol 1998 (Suppl 1); 27-35.
- Morovvati S, Nakagawa M, Sato Y, Hamada K, Higuchi I, Osame M. Phenotypes and mitochondrial DNA substitutions in families with A3243G mutation. Acta Neurol Scand 2002 Aug;106(2):104-8
- Silvestri G, Rana M, Odoardi F, et al. Singlefiber PCR in MELAS (3243) patients: correlations between intratissue distribution and phenotypic expression of the mtDNA(A3243G) genotype. Am J Med Genet 2000 Sep 18;94(3):201-6.

