Keywords
Crosscarmellose sodium; Sodium starch glycolate; Diclofenac sodium; Direct compression technique
Introduction
Oral drug delivery has been known for decades as the most widely utilized route of administration among all the routes that have been explored for the systemic delivery of drugs using different dosage forms [1]. In any case, numerous individuals particularly older and kids think that it’s hard to swallow the tablet which turns into a noteworthy disadvantage for their utilization. Late advances in novel drug delivery system (NDDS) aim to enhance safety and efficacy of drug molecules by formulating a convenient dosage form for administration and to achieve better patient compliance [2-5]. Solid dosage forms are popular because of low cost, ease of administration, accurate dosage self-medication, pain avoidance, and the most importantly the patient compliance. The most prevalent solid dosage forms are being tablets and capsules. One essential downside of such dosage forms is Dysphagia (trouble in gulping) is basic among all age gatherings. Normal grumblings about the trouble in gulping tablets are size, surface, and taste of tablets. Geriatric and paediatric patients and voyaging patients, who might not have prepared access to water, are most needing simple gulpinw g dosage forms. To satisfy these medicinal needs, pharmaceutical technologists have built up a novel oral dosage form known as orodispersible tablets (ODTs) which disintegrate quickly in salivation, normally inside only seconds, without the need to take water. Drug disintegration and absorption, just as beginning of clinical impact and drug bioavailability, might be fundamentally more noteworthy than those as contrasted and customary dosage forms. ODTs releases the medicament in the mouth for absorption through local oromucosal tissue and through pre-gastric (oral cavity, pharynx, and esophagus), gastric (stomach), and post-gastric (small and large intestine) segments of gastrointestinal tract [6,7].
Materials and Methods
We have used different excipients along with the Diclofenac sodium drug to prepare 8 formulations such as sodium starch glycolate, carboxy methyl cellulose, lactose, mannitol, croscarmellose sodium, micro crystalline cellulose, magnesium stearate and talc.
Drug profile
Diclofenac sodium
Diclofenac sodium is an NSAID (Non-Steroidal Anti- Inflammatory Drug) which is used as analgesic and antiinflammatory drug. It is freely soluble in methanol, soluble in ethanol (95%), sparingly soluble in water and in glacial acetic acid, practically insoluble in ether, in chloroform and in toluene. It is well absorbed following oral administration. It undergoes first pass metabolism, only 50-60% of a dose reaches systemic circulation as unchanged drug peak plasma concentration usually attained within about 1 hr (Diclofenac potassium conventional tablets), 2 hrs (Diclofenac sodium delayed release tablets). Absorbed into systemic circulation following topical administration as gel or transdermal system, plasma concentrations generally very low compared with oral administration [3,8-10].
Mechanism of action: The exact mechanism of action is not entirely known, but it is thought that the primary mechanism responsible for its anti-inflammatory, antipyretic and analgesic action is inhibition of prostaglandin synthesis by inhibition of cyclooxygenase (COX). It also appears to exhibit bacteriostatic activity by inhibiting bacterial DNA synthesis. Inhibition of COX also decreases prostaglandins in the epithelium of stomach, making it more sensitive to corrosion by gastric acid. This is also the main side effect of Diclofenac. It has low to moderate preference to block the COX-2 isoenzymes and is said to have, therefore a somewhat lower incidence of gastrointestinal complaints than noted with indomethacin and aspirin. The action of one single dose is much longer (6-8 hrs) than the very short half-life that the drug indicates. This could be partly because it persists for over 11 hrs in synovial fluid. Diclofenac may also be a unique member of NSAIDs [11]. There is some evidence that Diclofenac inhibits the lipoxygenase pathways, thus reducing formation of the leukotrienes. There is also speculation that Diclofenac may inhibit phospholipase-A, as part of its mechanism of action. These additional actions may explain the high potency of Diclofenac. It is the most potent NSAID on a broad basis. There are marked differences among NSAIDs in their selective inhibition of two types of cyclooxygenases, COX-1 and COX-2. Much pharmaceutical drug design has attempted to focus on selective COX-2 inhibition as a way to minimize the gastrointestinal side effects of NSAIDs like aspirin. In practice, use of some COX-2 inhibitors with their adverse effects has led to massive numbers of patients by heart attack, yet other significantly COX-selective NSAIDs such as Diclofenac have been well tolerated by most of the population. Besides the well-known and often cited COX inhibition, a number of other molecular targets of Diclofenac that could contribute to its pain relieving actions have recently been identified [12]. These include:
• Blockage of voltage dependant sodium channels (after enactment of the channel, Diclofenac restrains its reactivation otherwise called stage hindrance).
• Blockage of acid-sensing ion channels (ASICs).
• Positive allosteric balance of KCNQ (Potassium Channel, Voltage-Gated, KQT-like subfamily) and BK potassium channels (Diclofenac opens these channels, prompting hyper polarization of the cell layer).
Excipient profile
Croscarmellose sodium: Croscarmellose sodium stable through hygroscopic material. In tablets formulations, croscarmellose sodium might be utilized in both direct compression and wet granulation process. At the point when utilized in wet granulation, the croscarmellose sodium ought to be included both the dry and wet phases of the procedure (intra and additional granularly). Croscarmellose sodium at concentration up to 5% w/w may be used as a tablet disintegrant; although normally 2% w/w is used in tablet prepared by a wet granulation process [13].
Sodium starch glycolate: SSG is a practically white free streaming exceptionally hygroscopic powder. It is generally utilized in oral pharmaceutical as a crumbling in case and tablet. It is usually utilized in tablets arranged by either direct compression or wet-granulation forms. The standard concentration utilized in a formulation is somewhere in the range of 2% and 8%, with the ideal focus about 4%, in spite of the fact that by and large 2% is adequate. Disintegration occurs by rapid uptake of water followed by rapid and enormous swelling. [14].
Microcrystalline cellulose: Microcrystalline cellulose is a decent disintegrant (and a cover as well) yet at high compression drives, it might impede drug dissolution. One valuable excipient for direct compression is microcrystalline cellulose (MCC). Aside from its utilization in direct compression, microcrystalline cellulose is used as diluents in tablets arranged by wet granulation, as filler in capsules and for the creation of spheres. It is currently accessible in a wide assortment of evaluations; varying in parameters, for example, particle size, particle size distribution, density, and moisture. More up to date materials comprise of MCC co-prepared with different excipients. Therefore, there exists a scope of stream properties and compressibility's for MCC products that bring about contrasting tablet qualities and assembling requirements, which conceivably could influence drug dissolution [15].
Magnesium stearate: Magnesium stearate is an exceptionally fine, light white, hastened or processed, intangible powder of low mass thickness, having a black out smell of stearic acid and a characteristic taste. The powder is oily to contact and promptly holds fast to the skin. Magnesium stearate is generally utilized in beauty care products, nourishments, and pharmaceutical formulations. It is principally utilized as an ointment in case and tablet produce at focuses somewhere in the range of 0.25% and 5.0% w/w. It is likewise utilized in obstruction creams [16].
Talc: Powder is an extremely fine, white to greyish white, odourless, imperceptible, unctuous, crystalline powder. It sticks promptly to the skin and is delicate to contact and free from lumpiness. Powder assimilates inconsequential measure of water at 25°C and relative humidities up to 90%. Powder was once broadly utilized in oral solid dose formulations as a lubricant and diluent. It is generally utilized as a dissolution retardant in the advancement of controlled release products. Talc is additionally utilized as a lubricant in tablet formulations in a novel powder covering for broadened release pellets and as an adsorbent.
Sugar based excipients: This is another approach to manufacture ODT by direct compression. The use of sugarbased excipients is especially bulking agents like dextrose, fructose, isomalt, lactitol, maltitol, maltose, mannitol, sorbitol, starch hydrolysate, polydextrose, and xylitol which display high aqueous solubility and sweetness and hence impart taste masking property and a pleasing mouth feel. So we have used mannitol as taste masking agent.
Identification of drug
The drug was distinguished by melting point determination and ultraviolet spectroscopy (UV) [17].
Ultraviolet spectroscopy: The samples were subjected to UV spectrophotometric analysis and were scanned for absorption maxima (λmax) in the range of 200-400 nm using UV-Vis spectrophotometer in an appropriate medium. The obtained data was compared with that of reference values in literature [18].
Melting point: Melting point of Diclofenac was determined by capillary tube method. Fine powder of the drug was filled into a glass capillary tube which was previously sealed at one end. The capillary tube tied to a thermometer was subjected to increasing temperatures and the temperature at which Diclofenac melts was recorded [17].
Analytical methods for estimation of diclofenac sodium
Preparation of reagents
Preparation of phosphate buffer pH 6.8:
Preparation of sodium hydroxide solution (0.2 M): Sodium hydroxide pellets (8 g) were weighed and moved into a 1000 ml volumetric carafe. Adequate amount of distilled water was included and the pellets were dissolved. The volume was at last made up to 1000 ml with distilled water.
Preparation of potassium dihydrogen phosphate solution (0.2 M): Potassium dihydrogen phosphate (27.218 g) was weighes and moved into a 1000 ml volumetric flask. Adequate amount of distilled water was included and the substances were dissolved. The volume was made up to 1000 ml with distilled water.
Preparation of potassium dihydrogen phosphate solution (0.2 M): Potassium dihydrogen phosphate (27.218 g) was weighes and moved into a 1000 ml volumetric flask. Adequate amount of distilled water was included and the substances were dissolved. The volume was made up to 1000 ml with distilled water.
Construction of calibration curve of diclofenac sodium: Accurately weighed 25 mg of Diclofenac sodium was transferred into a 25 ml volumetric flask. It was dissolved in sufficient amount of distilled water and finally the volume was made up to 25 ml. This gives a solution having a concentration of 1 mg/ml Diclofenac sodium stock solution. From the primary stock, 1 ml was withdrawn and diluted to 10 ml with distilled water. From the secondary stock 0.5, 1, 1.5, 2, 2.5 and 3 ml was separately taken and individually made up to 10 ml with distilled water. The absorbance values were determined at 275 nm using UV spectrophotometer. A graph was plotted taking absorbance and concentration (Figures 1 and 2 and Table 1).
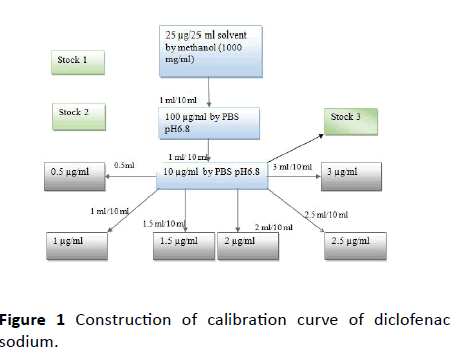
Figure 1 Construction of calibration curve of diclofenac sodium.
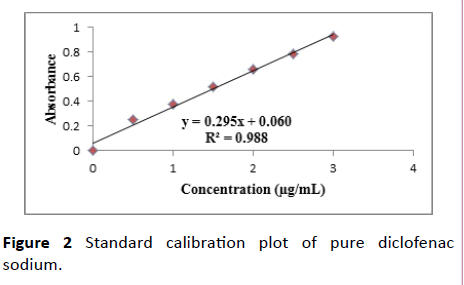
Figure 2 Standard calibration plot of pure diclofenac sodium.
| S. No |
Concentration (μg/ml) |
Absorbance |
| 1 |
0.0 |
0.000 |
| 2 |
0.5 |
0.250 ± 0.23 |
| 3 |
1.0 |
0.381 ± 0.67 |
| 4 |
1.5 |
0.519 ± 0.43 |
| 5 |
2.0 |
0.663 ± 1.23 |
| 6 |
2.5 |
0.784 ± 0.65 |
| 7 |
3.0 |
0.930 ± 0.69 |
Table 1 Calibration curve data for pure diclofenac sodium.
Methodology
Direct compression is used to define the process by which tablets are compressed directly from the powder blends of active ingredients and suitable excipients. No pre-treatment of the powder blends by wet or dry granulation is involved. Direct compression technique does not require the use of water or heat during the formulation procedure and is the ideal method for moisture and heat-labile medications. This is a process of compressing mixed powders into tablets without the need of intermediate granulating step.
This technique involves conventional equipment, commonly available excipients and a limited number of processing steps. High doses can also be accommodated and the final weight of tablet can easily exceed than that of other production methods [19]. Orodispersible dissolving tablets of Diclofenac sodium were prepared by direct compression method. All the ingredients were passed through #60mesh separately. Then the ingredients were weighed and mixed in geometrical order and compressed into tablets of 200 mg by using 10-station Rotary mini press tablet machine (Table 2).
| Ingredients (mg) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
| Diclofenac sodium (mg) |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
| Sodium starch glycolate (mg) |
10 |
20 |
10 |
20 |
-- |
-- |
-- |
-- |
| Croscaramellose sodium (mg) |
-- |
-- |
-- |
-- |
10 |
20 |
10 |
20 |
| Micro crystalline cellulose (mg) |
131 |
121 |
-- |
-- |
131 |
121 |
-- |
-- |
| Lactose (mg) |
-- |
-- |
127 |
115 |
-- |
-- |
129 |
117 |
| Mannitol (mg) |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
| Poly ethylene glycol (mg) |
-- |
-- |
-- |
-- |
-- |
-- |
-- |
-- |
| Carboxy methyl cellulose (mg) |
-- |
-- |
4 |
6 |
-- |
-- |
-- |
24 |
| Magnesium stearate (mg) |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
| Talc (mg) |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
| Total weight (mg) |
200 |
200 |
200 |
200 |
200 |
200 |
200 |
200 |
Table 2 Formulation details of different diclofenac ODTs.
Results and Discussion
The present study was undertaken to formulate orosidipersible tablet of Diclofenac tablets with view to deliver the drug in rapid manner in oral cavity. For this study, we have used sodium starch glycolate of 10% and croscarmellose sodium of 10% and 20%. Total 8 batches are formulated by varying the concentration of two different super disintegrants.
Evaluation of fast dissolving tablets
Pre compression parameters:
Micromeretics study: The Angle of repose of the formulation was in the scope of 21.65°-30.45°, which demonstrates great stream properties of the distinctive mixes. The Carr's index, Hausner's ratios were observed to be in the scope of 11.76-18.45 and 1.16-1.27 demonstrating great compressibility.
The pre-compression parameters, for example, Angle of repose, tapped density, mass denisty, Hausner's ratio, and compressibility record are inside the limit demonstrating the agreeableness of the product to formulate into a dosage form. Orodispersible tablets were formulated by direct compression method by using 6 station tablet compression machine.
The prepared tablets were evaluated for post compression parameters such as weight variation, disintegration time, dissolution method, friability, hardness, and all the formulations are within the pharmacopoeial limit indicating the acceptability of the product in this dosage form (Table 3).
| Formulations |
Angle of repose
(Mean ± SD) |
Bulk density
(g/cc)
(Mean ± SD) |
Tapped density (g/cc)
(Mean ± SD) |
Carr’s Compressibility index |
Hausner’s ratio |
| F1 |
30.45 ± 0.54 |
0.55 ± 0.02 |
0.73 ± 0.05 |
18.45 |
1.22 |
| F2 |
26.43 ± 1.32 |
0.37 ± 0.04 |
0.56 ± 0.43 |
15.43 |
1.17 |
| F3 |
24.73 ± 1.04 |
0.48 ± 0.13 |
0.52 ± 0.08 |
14.87 |
1.12 |
| F4 |
21.65 ± 1.52 |
0.42 ± 0.04 |
0.64 ± 0.14 |
11.76 |
1.14 |
| F5 |
27.32 ± 1.33 |
0.46 ± 0.14 |
0.53 ± 0.02 |
12.01 |
1.14 |
| F6 |
26.24 ± 0.67 |
0.41 ± 0.01 |
0.64 ± 0.18 |
13.79 |
1.19 |
| F7 |
27.44 ± 0.68 |
0.44 ± 0.24 |
0.57 ± 0.09 |
14.65 |
1.21 |
| F8 |
28.62 ± 0.76 |
0.48 ± 0.16 |
0.62 ± 0.05 |
16.44 |
1.16 |
Table 3 Precompression parameters data for the selected batch formulation blend.
Post compression parameters:
Weight variation: All tablets from every formulation passed weight variation test, as the % weight variation under 7.5%, which was within the pharmacopoeial limits.
Friability: The friability of the formulations was observed to be between 0.33-0.75% and was inside the official prerequisite (i.e., less than 1%).
Hardness: The hardness was maintained to be within 2.8-3.2 kg/cm2, no variation in the hardness was found which clearly indicates that the blending was uniform.
In vitro dispersion time: The values of the formulations were found to be within the range of 31.2 seconds to 52.6 seconds. The formulation containing crosscarmellose sodium (CCS) (F6) showed a faster dispersion time of 31.3 seconds when compared with other formulations (Figure 3).

Figure 3 Picture showing dispersion of prepared ODTs.
In vitro disintegration time: In vitro disintegration time for all the formulations varied from 20.6-39.3 seconds. The formulation F6 showed better disintegration time of 20.6 seconds. All the formulations are disintegrated within 1 min indicating the suitability of product (orodispersible criteria) (Table 4).
| Formulation Code |
In vitro dispersion time (sec) |
In vitro disintegration time (sec) |
| F1 |
44.3 |
32.4 |
| F2 |
45.4 |
39.3 |
| F3 |
42.7 |
34.6 |
| F4 |
36.5 |
26.8 |
| F5 |
52.6 |
30.5 |
| F6 |
31.3 |
20.6 |
| F7 |
36.3 |
26.4 |
| F8 |
38.4 |
28.4 |
Table 4 In vitro evaluation of prepared ODTs.
In vitro drug release: Among all the formulations F1, F5 and F6 showed maximum drug release within 30 min compared to other formulations. F1 formulation has sodium starch glycolate (SSG) 10% as super disintegrant and F5 formulation has CCS (10%) and F6 formulation as CCS (20%).
By observing these 3 formulations drug release CCS is more suitable for ODT of Diclofenac sodium when compared with SSG. Formulation F6 containing superdisintegrant CCS (20%) gave 100% drug release compared to F5 formulation which is 82.3% drug release within 30 min.
Even though all the formulations compiling the quality controlling as per I.P the selection of formulation was done especially by considering the 100% drug release within 30 min. By considering this criterion, F6 formulation was selected as best formulation and was compared with the marketed formulation (Table 5 and Figure 4).
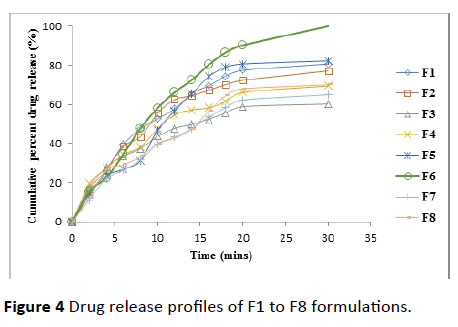
Figure 4 Drug release profiles of F1 to F8 formulations.
| Time (min) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
| 0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| 2 |
13.7 ± 0.25 |
16.6 ± 0.98 |
17.8 ± 0.58 |
19.4 ± 0.15 |
14.6 ± 0.35 |
15.5 ± 0.14 |
11.3 ± 0.47 |
12 ± 0.47 |
| 4 |
26.7 ± 0.32 |
26.7 ± 0.84 |
27.9 ± 0.38 |
27.4 ± 0.54 |
23.7 ± 0.34 |
22.6 ± 0.12 |
22.1 ± 0.41 |
26.7 ± 0.854 |
| 6 |
39.6 ± 0.32 |
38.6 ± 0.35 |
33.6 ± 0.38 |
34.4 ± 0.54 |
26.9 ± 0.14 |
34.7 ± 0.14 |
26.5 ± 0.21 |
29.2 ± 0.89 |
| 8 |
47.6 ± 0.35 |
43.6 ± 0.65 |
37.5 ± 0.24 |
38.3 ± 0.45 |
31.4 ± 0.12 |
47.9 ± 0.24 |
31.8 ± 0.12 |
33.4 ± 0.47 |
| 10 |
52.7 ± 0.32 |
55.5 ± 0.41 |
43.7 ± 0.39 |
47.5 ± 0.14 |
46.5 ± 0.74 |
58.2 ± 0.14 |
39.7 ± 0.31 |
40.1 ± 0.89 |
| 12 |
57.8 ± 0.31 |
62.5 ± 0.24 |
47.8 ± 0.41 |
54.8 ± 0.21 |
56.8 ± 0.45 |
66.3 ± 0.47 |
43.3 ± 0.13 |
42.8 ± 0.45 |
| 14 |
65.7 ± 0.87 |
64.5 ± 0.58 |
49.8 ± 0.38 |
57.2 ± 0.32 |
65.4 ± 0.47 |
72.5 ± 0.84 |
47.6 ± 0.01 |
47.3 ± 0.14 |
| 16 |
69.6 ± 0.65 |
67.3 ± 0.68 |
52.5 ± 0.21 |
58.3 ± 0.31 |
74.3 ± 0.14 |
80.3 ± 0.79 |
53.7±0.21 |
56.5 ± 0.12 |
| 18 |
74.6 ± 0.41 |
70.2 ± 0.28 |
55.8 ± 0.37 |
61.5 ± 0.14 |
79.1 ± 0.17 |
86.7 ± 0.47 |
58.4 ± 0.45 |
64.7 ± 0.47 |
| 20 |
77.5 ± 0.27 |
72.4 ± 0.98 |
58.9 ± 0.32 |
66.2 ± 0.14 |
80.7 ± 0.12 |
90.3±0.78 |
62.3 ± 0.85 |
68.02 ± 0.41 |
| 30 |
80.7 ± 0.28 |
77.2 ± 0.64 |
60.3 ± 0.14 |
69.5 ± 0.25 |
82.3 ± 0.15 |
100.02±0.41 |
65.2 ± 0.48 |
70.1 ± 0.47 |
Table 5 Drug release profiles of all prepared ODTs batches.
FTIR spectroscopy
FTIR investigation of pure drug, CCS and streamlined formulation (F6) containing Diclofenac sodium and CCS were obtained. IR spectra of Diclofenac sodium displayed a characteristic peak at 3384.80 cm-1 because of NH extending of the secondary amine, 1575.75 cm-1 attributable to –C=O extending of the carboxyl particle and at 747.66 cm-1 in view of C-Cl extending. Infrared spectra of enhanced formulation demonstrated the characteristic pinnacles of the unadulterated drug Diclofenac sodium. From the above elucidation, it was found that there is no shifting in the frequencies of above said functional groups. Thus no association among drug and excipients was found [20,21] (Figures 5 and 6).
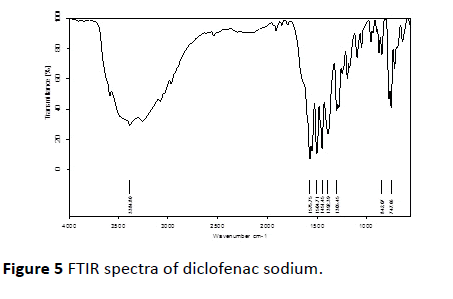
Figure 5 FTIR spectra of diclofenac sodium.
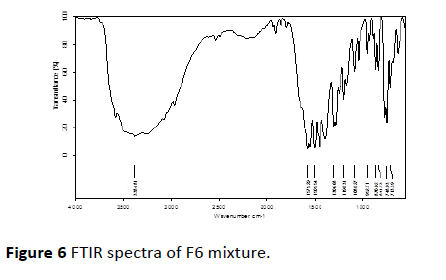
Figure 6 FTIR spectra of F6 mixture.
Comparative evaluation/similarity and dissimilarity factor evaluation
The outcomes were appeared in the Table 6 and Figure 7. The estimation of F1 and F2 were observed to be 5.99 and 53.26 individually. As the F1<15 and F2>50 shows similitude of drug release as that of marketed product.
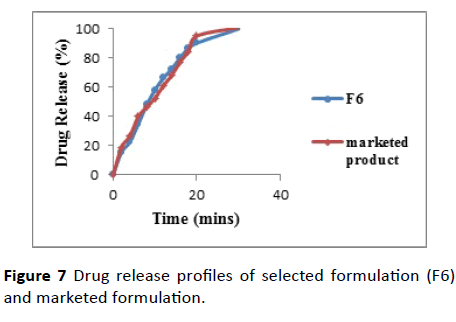
Figure 7 Drug release profiles of selected formulation (F6) and marketed formulation.
| Time (mins) |
F6 (selected formulation) (Tj) |
Marketd formulation (Rj) |
Tj-Rj |
[Tj-Rj]2 |
| 0 |
0.0 |
0.0 |
0.0 |
0.0 |
| 2 |
15.5 |
18.9 |
3.4 |
11.56 |
| 4 |
22.6 |
26.6 |
4.1 |
16.12 |
| 6 |
34.7 |
39.9 |
5.2 |
27.04 |
| 8 |
47.9 |
46.4 |
1.5 |
2.25 |
| 10 |
58.2 |
52.5 |
5.7 |
32.49 |
| 12 |
66.3 |
61.1 |
5.2 |
27.04 |
| 14 |
72.5 |
68.2 |
4.3 |
8.49 |
| 16 |
80.3 |
7703 |
3.2 |
6.64 |
| 18 |
86.7 |
84.5 |
2.2 |
4.84 |
| 20 |
90.3 |
95.1 |
4.8 |
23.04 |
| 30 |
100.02 |
101 |
0.9 |
0.92 |
| -- |
∑= 675.02 |
∑=671.5 |
∑=40.28 |
∑=1,622.47 |
Table 6 Comparative similarity and dissimilarity evaluation of selected formulation (F6) with that of marketed formulation.
The comparative drug release profiles of F6 and marketed tablets indicating that the release profiles of these 2 products are similar.
Hence F6 tablet formulated by employing CCS 20% as superdisintegrant and mannitol as diluent are considered to the best Diclofenac ODTs formulated and suitable for daily usage.
Conclusion
The present study was undertaken to formulate orosidipersible tablet of Diclofenac tablets with a view to deliver the drug in rapid manner in oral cavity. For this study, we have used sodium starch glycolate and croscarmellose sodium (10% and 20%). Total 8 batches are formulated by varying the concentrations of these two superdisintegrants. The pre compression parameters such as angle of repose, tapped density, bulk density, Hausner’s ratio, compressibility index are within the limit indicating the acceptability of the product to formulate into a dosage form. Orodispersible tablets are formulated by direct compression method by using 6 station tablet compression machine. The prepared tablets were evaluated for post compression parameters such as weight variation, disintegration time, dissolution method, friability, hardness, and all the formulations are within the pharmacopoeia limit indicating the acceptability of the product in this dosage form Kakade [22]. All the formulations are disintegrated within 1 min indicating the suitability of product (orodispersible criteria). Among all the formulations F1, F5 and F6 showed maximum drug release within 30 min compared to other formulations. F1 formulation contains sodium starch glycolate (SSG) 10% as super disintegrant and F5 formulation has crosscarmellose sodium (CCS) 10% and F6 of 20%. By observing these 3 formulations drug release CCS is more suitable for ODT of Diclofenac sodium compared with SSG. F6 formulation gave 100% drug release compared to F5 formulation which is 82.3% drug release within 30 min. Even though all the formulations compiling the quality controlling as per I.P the selection of formulation was done especially by considering the 100% drug release within 30 min, in this F6 formulation was selected as best formulation and was compared with the marketed formulation revealed that no interaction of drug and excipients used. This study concluded that for Diclofenac drug, crosscarmellose sodium of 20% and diluent microcrystalline cellulose is more suitable as ODTs and showing similar drug release as that of marketed Diclofenac product. Hence this selected formulation (F6) was more suitable for daily usage.
Acknowledgements
We express our sincere thanks to Dr. Y. Srinivasa Rao, Principal of Vignan Instistute of Pharmaceutical Technology for his valuable guidance throughout the research work and we also express our profound sense of gratitude to our project guide Dr. B. Bhavani, Associate professor, Vignan Institute of Pharmaceutical Technology for her valuable guidance, constant encouragement and thought provoking comments during the course of this work.
Conflict of Interest
The authors declare no conflict of interests.
24055
References
- Guhmann M, Preis M, Gerber F, Pöllinger N, Breitkreutz J, et al. (2015) Design, development and in-vitro evaluation of diclofenac taste-masked orodispersible tablet formulations. Drug Dev Ind Pharm 41(4): 540-541.
- Amit M, Vandana S, Arun G, Ashish A (2012) Formulation and evaluation of fast dissolving tablets of diclofenac sodium using different super disintigrants by direct compression method. IJPBA 3(4): 1003-1007.
- Sudhir B (2011) A novel approach for drug delivery: Fast dissolving tablet of diclofenac sodium. AJBPR 3(1): 606-616.
- Karthikeyan M, Umarul MA, Megha M, Shadeer HP (2012) Formulation of diclofenac tablets for rapid pain relief. Asian Pac J Trop Biomed 2(1): 308-311.
- Priyanka K, Vikesh S (2014) Formulation and evaluation of fast dissolving tablets of diclofenac sodium using PVP. IJPRR 3(7): 12-19.
- El-Setouhy DA, El-Malak NS (2010) Formulation of a novel tianeptine sodium orodispersible film. Aaps Pharmscitech 11(3): 1018-1025.
- Khokhar P, Shukla V (2014) Formulation and evaluation of fast dissolving tablets of diclofenac sodium using PVP. IJPRR 7: 12-19.
- Menasse R, Hedwall PR, Kraetz J, Pericin C, Riesterer L, et al. (1978) Pharmacological properties of diclofenac sodium and its metabolites. Scand J Rheumatol 7(22): 5-16.
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, et al. (2000) Gastrointestinal toxicity with celecoxib vs. nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. JAMA 284(10): 1247-1255.
- Singh G (1998) Recent considerations in nonsteroidal anti-inflammatory drug gastropathy. Am J Med 105(1): 31-38.
- Ferrero C, Munoz N, Velasco MV, Muñoz-Ruiz A, Jiménez-Castellanos R (1997) Disintegrating efficiency of croscarmellose sodium in a direct compression formulation. Int J Pharm 147(1): 11-21.
- Chaulang G, Patel P, Hardikar S, Kelkar M, Bhosale A, et al. (2009) Formulation and evaluation of solid dispersions of furosemide in sodium starch glycolate. Trop J Pharm Res 8(1): 43-51.
- Battista OA, Smith PA (1962) Microcrystalline cellulose. Ind Eng Chem Res 54(9): 20-29.
- Vromans H, Lerk CF (1988) Densification properties and compactibility of mixtures of pharmaceutical excipients with and without magnesium stearate. Int J Pharm 46(3): 183-192.
- Giordano F, Rossi A, Pasquali I, Bettini R, Frigo E, et al. (2003) Thermal degradation and melting point determination of diclofenac. J Therm Anal Calorim 73(2): 509-518.
- Sena MM, Chaudhry ZF, Collins CH, Poppi RJ (2004) Direct determination of diclofenac in pharmaceutical formulations containing B vitamins by using UV spectrophotometry and partial least squares regression. J Pharm Biomed Anal 36(4): 743-749.
- Guhmann M, Preis M, Gerber F, Pöllinger N, Breitkreutz J, et al. (2015) Design, development and in-vitro evaluation of diclofenac taste-masked orodispersible tablet formulations. Drug Dev Ind Pharm 41(4): 540-551.
- Smith BC (2011) Fundamentals of Fourier transform infrared spectroscopy. CRC Press p: 207.
- Davies MC, Binns JS, Melia CD, Bourgeois D (1990) Fourier transform Raman spectroscopy of polymeric biomaterials and drug delivery systems. Spectrochim Acta A Mol Biomol Spectrosc 46(2): 277-283.
- Kakade SM (2016) Formulation and evaluation of mouth dissolving tablets of losartan potassium by direct compression techniques. Int J Res Pharm Sci 1(3): 290-295.