Keywords
Neurodegeneration,DNA damage and repair, Purα, Cell cycle
Introduction
Neurodegeneration is the common feature of many diseases of the central nervous system (CNS) [1-3], most notably in Alzheimer’s disease (AD) [4]. However, the molecular mechanism underlying the development and progression of the disease is poorly understood [5-7]. Deficiency in the processes controlling DNA repair and genomic integrity is among the potential biological events that can influence neuronal cell survival and differentiation under normal cell conditions, and its degeneration may contribute to the development of the disease [3]. In postmitotic neurons, breaks occurring in DNA are mainly removed by the single-strand break repair (SSBR) pathway [8], or, in the case of double strand breaks (DSB), by non-homologous end-joining (NHEJ) pathway [9]. There are several cellular factors including PARP-1 [10], DNA ligase [11], polynucleotide kinases [12], Ku70 and Ku80 [13,14], whose function in DNA repair have been well studied. The ataxia telangiectasia (AT) mutated (ATM) kinase, which plays a role as a sensor for DSB [15] and activates DNA repair and signaling for cell cycle checkpoints in G1, S, and G2, has drawn much attention as a key player in the control of genomic stability, apoptosis and cell survival [16,17].
Among the recently discovered cellular proteins that have captured people’s attention is Purα whose role in neuronal cell survival and differentiation has been demonstrated in an animal model. As a transcriptional factors, Purα could bind to the purine-rich sequences of DNA in a special way by which it can identify the specific sequences like (GGN) in the DNA sequences [18]. The model of Purα knock out transgenic mice has been successfully established and it provided a perfect model for the research of the biological functions of Purα in many disciplines [19]. The Purα deficient mice exhibited noticeable neurological defects and showed severe neuronal cell abnormalities with fewer neurons presented in regions of the hippocampus and the cerebellum [19]. It is interesting that another transgenic Purα knock out mouse model created by Hokkanen et al. [20] is not consistent with the results reported by Khalili et al. [19], they disputed the results reported by Khalili et al. and they reported that lack of Purα prolonged the postnatal proliferation of neuronal precursor cells both in the hippocampus and in the cerebellum, however, without affecting the overall number of post-mitotic neurons. Independent of these findings, they also declared that they observed alterations in the expression and distribution of the dendritic protein MAP2, the translation of which has been proposed previously to be Purα-dependent. Both of these two research groups observed the phenomenon that mice lack of Purα generated a continuous tremor which persisted throughout lifetime. But the most important is that they reported in their model that Purα-/- mice displayed a megalencephaly and histopathological findings including axonal swellings and hyperphosphorylation of neurofilaments, but not as reported by Khalili research group that the Purα-/- mice would die at the fourth week after birth. It was hard to tell why the two groups presented the different research results on Purα knock out mice, but in Hokkanen S’ paper, although they presented plentiful immunohistochemical pictures to affirm their findings in Purα-/- mice brain, but the most fatal weakness of this paper was that they did not present any western blotting result to prove that Purα gene has really been knocked out and this is the Achilles’ heel of their work. Perhaps it was just a truncated knock out model of Purα.
Purα is essential for the control of cell growth and may play an important role in maintenance of the genomic integrity in central nervous system [21,22]. The mutations in Purα caused a profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome [23,24]. So it is important to understand the roles that Purα plays in the central nervous system, especially the function of Purα in maintenance of the stability of genomic DNA and to clarify the association with neurodegenerative diseases.
In this review, we present an overview of the current understanding of the molecular basis for neuronal DNA repair deficiencies associated with neurodegeneration. And also, the function of human transcriptional activator, Purα, in the maintenance of stability of genomic integrity in the central nervous system was also discussed in this mini review.
Neurodegeneration and DNA damage
Neurodegeneration is the common hallmark of many nervous system and aging diseases, such as Ataxia Telangiectasia (AT) [25], Nijmegen breakage syndrome (NBS) [26], Huntington Disease (HD) [27], Parkinson’s disease (PD) and Alzheimer’s disease (AD) [28]. Furthermore, deficiency in repair of nuclear and mitochondrial DNA damage has been linked to several neurodegenerative disorders. Many recent experimental results indicate that the post-mitotic neurons are particularly prone to accumulation of unrepaired DNA lesions and potentially lead to progressive neurodegeneration. All of these suggest that maintenance of genomic stability is critical for neuronal development and functions [29-31]. DNA damage induced diseases in the nervous system can be caused by mutations in the genes involved in the DNA damage response, and a number of these genes are required for the normal development of the nervous system. Lalani et al. reported that human transcriptional activator Purα, which is considered as one of cellular factors closely associated with nervous development, its mutation results in 5q31.3 microdeletion syndrome, which is characterized by neonatal hypotonia, encephalopathy with or without epilepsy and severe developmental delay, and the minimal critical deletion interval harbors three genes. They described 11 individuals with clinical features of 5q31.3 microdeletion syndrome and de novo mutations in PURA, encoding transcriptional activator protein Purα, within the critical region. These data implicated causative PURA mutations responsible for the severe neurological phenotypes observed in this syndrome [23]. The research by Hunt et al. with whole exome sequencing in family trios revealed de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability, further confirmed that these findings provided definitive evidence for the role of PURA in causing a variable syndrome of neurodevelopmental delay, learning disability, neonatal hypotonia, feeding difficulties, abnormal movements and epilepsy in humans, and help clarify the role of PURA in the previously described 5q31.3 microdeletion phenotype [24]. Combining the results of Purα knock out mice and the findings in 5q31.3 microdeletion syndrome, it is not difficult to conclude that the mutation of the factors associated with nervous development would disturb the stabilities of genomic integrity and cause the severe defects in nervous system.
After completing the normal development processes, neurons enter a post-mitotic state and stay in a resting status. When damaged genomic DNA was not completely repaired or the ability of repair was deficiency, the unrepaired DNA would be accumulated in the neuron which will be provoked to reenter into cell cycle. When post-mitotic neuron reentered into cell cycle, since they lacked the necessary cellular cycle proteins such as relative cyclins and CDKs, it could not complete the all cell cycles. Reactivation of the cell cycle in neurons would lead to apoptosis. Neurons are coming out from G0 to G1, at the G1/S checkpoint, neurons are either coming back in G0 by differentiation or entering apoptotic process. At these circumstances, they expressed a few markers including cyclin D1 and E2F-responsive gene products. The phenomenon of reactivation of the cell cycle in neurons was reported in Alzheimer’s disease, neurons are coming out from G0 to G1, both G1/S and G2/M checkpoint markers have been found in neurofibrillary tangles which suggests that neurons bypass the classical neuronal apoptosis [32] (Figure 1). The association between the cell cycle activity and DNA damage have been observed in neurons in association with various neurodegenerative conditions [33]. While there is strong evidence for a causative role for these events in neurotoxicity, it is unclear how they are triggered and why they are toxic. Generally speaking, the two events may be triggered in common by deregulation of fundamental processes, such as chromatin modulation, which are required for maintaining both DNA integrity and proper regulation of cell cycle gene expression [33].
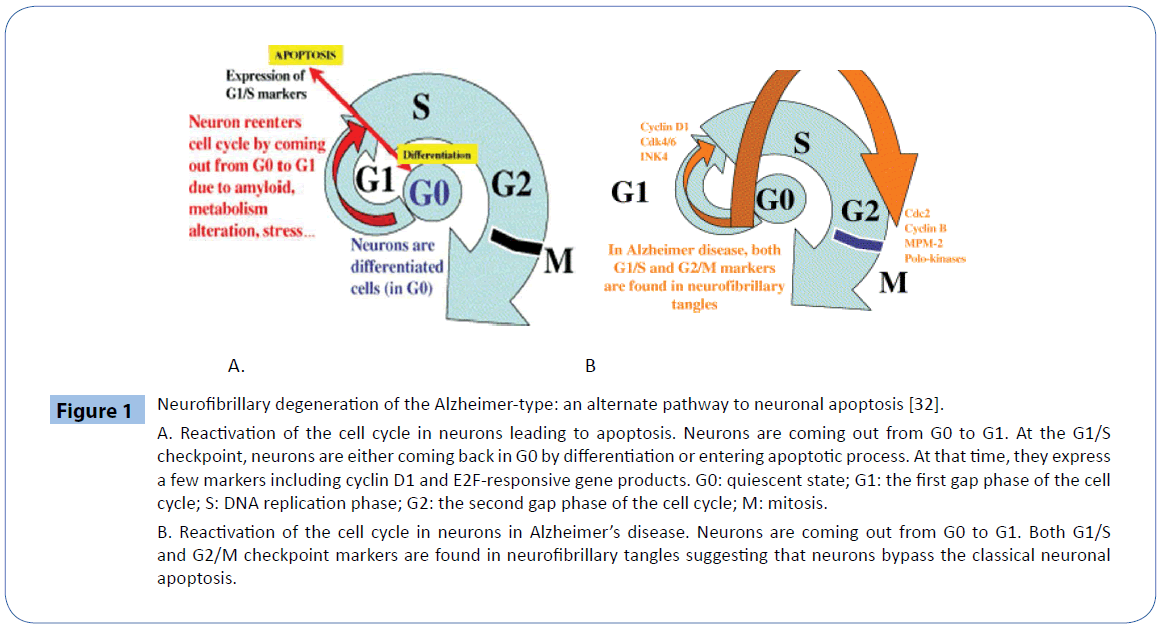
Figure 1: Neurofibrillary degeneration of the Alzheimer-type: an alternate pathway to neuronal apoptosis [32].
A. Reactivation of the cell cycle in neurons leading to apoptosis. Neurons are coming out from G0 to G1. At the G1/S checkpoint, neurons are either coming back in G0 by differentiation or entering apoptotic process. At that time, they express a few markers including cyclin D1 and E2F-responsive gene products. G0: quiescent state; G1: the first gap phase of the cell cycle; S: DNA replication phase; G2: the second gap phase of the cell cycle; M: mitosis.
B. Reactivation of the cell cycle in neurons in Alzheimer’s disease. Neurons are coming out from G0 to G1. Both G1/S and G2/M checkpoint markers are found in neurofibrillary tangles suggesting that neurons bypass the classical neuronal apoptosis.
Re-entry of post-mitotic neurons into the cell cycle and the presence of unrepaired DNA damage can induce apoptosis, lead to neuronal degeneration [31,34,35]. Amyloid beta peptide (Aβ) and tau are two characteristic lesions in the brains of AD patients. However, Aβ induces oxidative stress and stimulates neuron to reenter into the cell cycle [31]. The developing nervous system is highly susceptible to DNA damage-induced apoptosis. All these observations suggested that DNA repair plays an important role in the normal development and maintenance the stabilities of the demonic integrity of the nervous system [29,36,37]. Therefore, information regarding the repair activities of cells of the nervous system appears necessary for our mechanistic understanding of several related diseases.
In addition, it has been reported that terminally differentiated neurons only repair the genes expressed by themselves and they have lost the functions of global genomic repair [38,39]. Also in the aging cortex, the ability of base-excision repair is reduced, which leads to DNA damage in the promoters of genes with reduced expression [40], and also, there are reports that non-homologous end joining (NHEJ) has been reduced in the differentiated PC12 cells [21]. These results suggest that the effective treatment measures for several relevant diseases of CNS could be developed only after the operation of specific regulatory mechanisms for DNA maintenance in neural cells was understood.
DNA repair and neurodegeneration
The integrity of our genetic materials is under constant attack from numerous exogenous endotoxins, including ionizing radiation (IR), UV light and chemotherapeutic agents, as well as endogenous processes associated with oxidative metabolism, stalled DNA replication and V(D)J recombination. All these exogenous and endogenous genotoxic agents can continuously generate DNA single-strand breaks (SSB) and double strand breaks (DSB). The consequence of defective DNA damage response are well studied in proliferating cells, especially with regards to the development of cancer, yet its precise role in the nervous system are relatively poorly understood.
Among the fundamental processes, crucial for viability of organisms, including humans, are appropriate cellular signaling response to DNA damage and the ability to repair such damage. To protect against this damage all cells have various DNA repair pathways. The four major pathways for repairing damage to bases are nucleotide excision repair (NER), base excision repair (BER), mismatch repair (MMR) and double–strand break repair (DSBR) (Figure 2). NER excises bulky helix-distorting DNA lesions and BER repairs damage to a single nucleotide base, whereas MMR corrects mismatches of the normal bases; such as failure to maintain normal Watson–Crick base pairing. Breakage of the DNA backbone also occurs, either in the form of a singlestrand break (SSB) or a double-strand break (DSB). SSBs are handled by the BER pathway. The repair of DNA DSBs involves one of two mechanisms: non-homologous end joining (NHEJ) or homologous recombination (HR). NHEJ directly joins the broken ends, whereas HR uses the intact sister chromatid as a template for repair. In addition, a type of repair termed direct reversal (DR) can reverse some forms of base damage without removing the base. Translesion DNA synthesis (TLS) uses specialized DNA polymerases to replicate past lesions in the DNA, which although more error-prone than BER, NER and MMR, may reduce the immediate danger of DSBs [41].
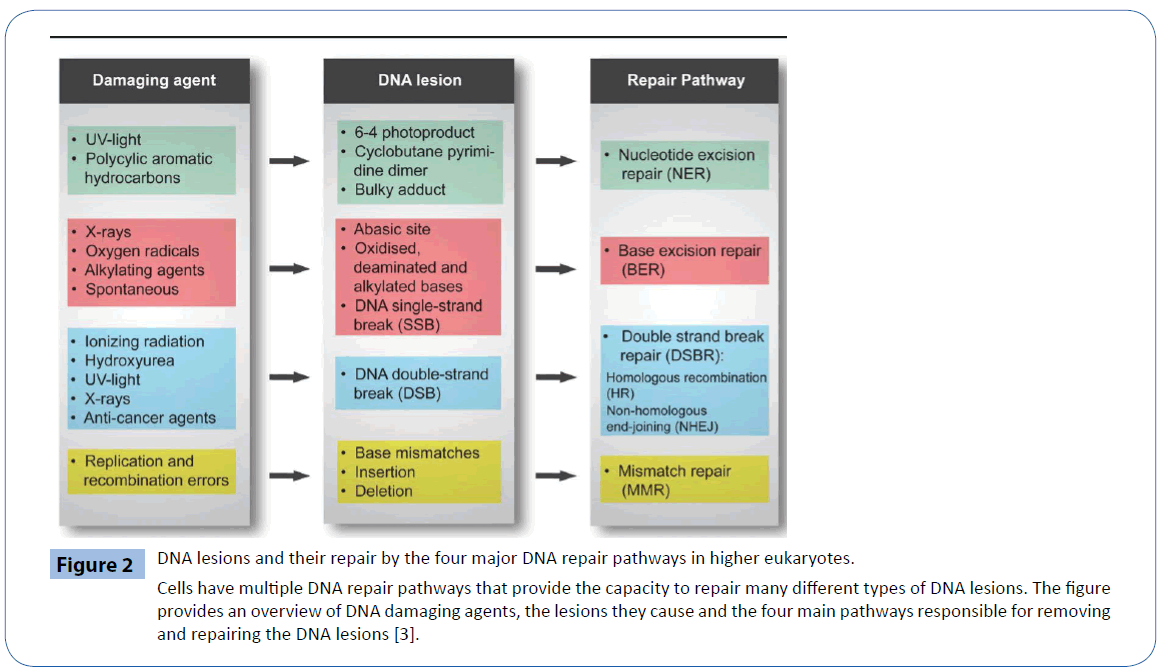
Figure 2: DNA lesions and their repair by the four major DNA repair pathways in higher eukaryotes.
Cells have multiple DNA repair pathways that provide the capacity to repair many different types of DNA lesions. The figure provides an overview of DNA damaging agents, the lesions they cause and the four main pathways responsible for removing and repairing the DNA lesions [3].
In response to DNA damage, cells activate several pathways of the DNA damage response including DNA repair, cell cycle checkpoints, transcription and apoptosis [42,43]. Damaged DNA is sensed either by the ATM or ATR proteins to arrest the progression of cell cycle in G1, S or G2 phase. These checkpoints are regulated by the Chk1 or Chk2 that inhibit the activation of CDC25A or CDC25C and cause the inactivation of CDK/Cyclin complex. Depending on the nature of damage, DNA lesions can be removed by several DNA repair mechanisms as described above. Double strand break (DSB) is repaired either by homologous recombination repair (HRR) or non-homologous end-joining (NHEJ). If unrepaired or if misrepaired, DSB can cause genomic instability which leads to cancer, genetic diseases and premature aging [42, 44-47]. Repair by homologous recombination requires extensive homology and RAD52 epitasis group of genes (RAD50- 55), XRCC2 and XRCC3 [48-50]. NHEJ does not require homology and is greatly facilitated by the DNA dependent protein kinase (DNA-PKs), Ku and ligase IV/XRCC4 complex [51-53]. NBS1 forms a complex with MRE11 and Rad50 called the MRN complex, which plays a role in both NHEJ and HRR. ATM and ATR, act as a sensors of DNA damage to induce specific signal transduction pathways. BRCA1 and BRCA2 are also involved in homologous recombination repair (HRR) [54-56]. Recent studies show that NHEJ and HRR are probably also functioning in a coordinated manner to repair DSB [57]. Genetic studies have demonstrated that deficiency of DNA repair protein results in neurodegeneration. MRE11 is reduced in neurons of the cortex of patients suffering from Alzheimer’s disease (AD), which suggest that loss of MRE11 will be associated with the pathogenesis of AD [38]. That deletion of DSB repair proteins, such as Ku, XRCC4, DNA ligase IV and XRCC2, leads to neuronal apoptosis suggest that DSB repair is critical for the stability of the nervous system [58-61]. However, ablation of p53 or ATM restores neuronal development in mice deficient in DNA repair proteins [62-65]. These observations suggest that DSB repair, particularly NHEJ, plays a critical role in the maintenance of genomic integrity in the nervous system. The SSBR complex includes poly(ADP-ribose) polymerase 1(PARP-1), XRCC1, DNA ligase III, polynucleotide kinase (PNK) and DNA polymerase β. Hereditary spin-cerebellar ataxia has been associated with mutation in genes involved in SSBR [66]. Since the DNA replication in post-mitotic neurons ceased, DNA damages are repaired predominantly by SSB and NHEJ in the nervous system (Figure 3).
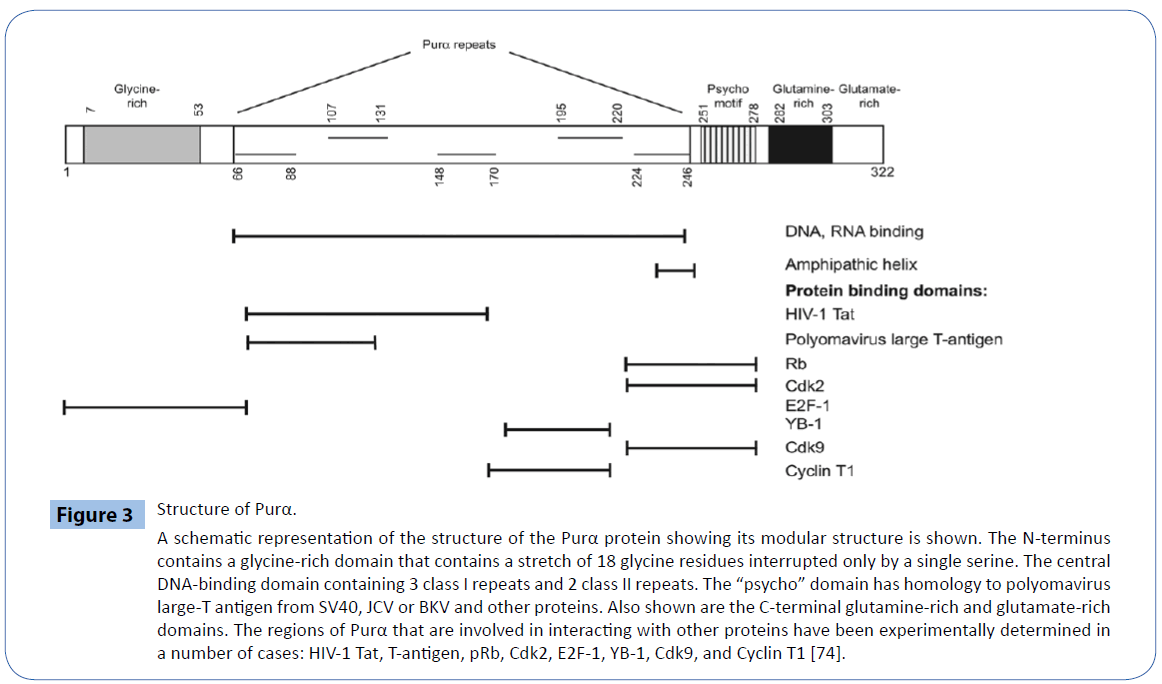
Figure 3: Structure of Purα.
A schematic representation of the structure of the Purα protein showing its modular structure is shown. The N-terminus contains a glycine-rich domain that contains a stretch of 18 glycine residues interrupted only by a single serine. The central DNA-binding domain containing 3 class I repeats and 2 class II repeats. The “psycho” domain has homology to polyomavirus large-T antigen from SV40, JCV or BKV and other proteins. Also shown are the C-terminal glutamine-rich and glutamate-rich domains. The regions of Purα that are involved in interacting with other proteins have been experimentally determined in a number of cases: HIV-1 Tat, T-antigen, pRb, Cdk2, E2F-1, YB-1, Cdk9, and Cyclin T1 [74].
The role of Purα in DNA damage and repair
Purα is a ubiquitous nucleic acid-binding protein that has been originally purified from the mouse brain based on its ability to bind to a DNA sequence derived from the promoter of the mouse myelin basic protein gene [67,68]. Human Purα is characterized by the ability to bind to a DNA sequence present upstream of the human c-myc gene, its cDNA has been cloned from HeLa cells and sequenced already [69,70]. The sequence of mouse Purα [71] and human Purα [69] are nearly identical with only 2 out of 322 amino acids residues differed. The DNA-binding domain of Purα is strongly conserved throughout evolution. Purα is a member of Pur family of proteins along with Purβ and Purγ, for which two isoforms exist; that arise from the usage of alternative polyadenylation sites. Purα is expressed virtually in every metazoan tissue and it is a multifunctional protein that can bind to both DNA and RNA and functions in the initiation of transcription, DNA replication, DNA repair and RNA transport [18,72]. Purα associates with DNA sequences that are close to viral and cellular origins of replication (For the detail functions and genetic information of Purα, please refer to the review by Gordon J, et al. [73] and White Mk, [74]. Since initiation of transcription and replication requires unwinding of duplex DNA, this is consistent with evidence that Purα possesses DNA helixdestabilizing activity [75]. Several lines of evidence suggest that Purα is a major player in regulation of the cell cycle and oncogenic transformation. Purα binds to several cellular regulatory proteins including the retinoblastoma protein (pRB) [76], E2F1 [77], Sp1 [78], YB1 [79] and RhoA [80]. Purα also binds to the large T antigen of JCV (T-Ag) [81]. The intracellular level of Purα varies during the cell cycle, declining at the onset of S-phase and peaking during mitosis [82] and G2/M checkpoints [83]. When expressed in Rastransformed NIH-3T3 cells, Purα inhibits their ability to grow in soft agar [84]. Ectopic expression of Purα suppresses the growth of several tumor cell lines including glioblastoma-origin cells [85]. The growth-inhibitory effects of Purα are supported by the observation that gene expression in chronic myeloid leukemia patients is down-regulated by Purα. In addition, it has been reported that there are deletions of Purα in the myelodysplastic syndrome, a condition that can progress to acute myelogenous leukemia, a result consistent with a haploinsufficiency role of Purα in protecting against neoplasia. Thus Purα is an important transcriptional factor that exerts a key role in the regulation of cell proliferation.
Purα knockout (Purα-/-) mice were genetically engineered. The mice were normally at birth, but at the 15th postnatal day they develop neurological problems with severe tremor and spontaneous seizures; they die by the 4th week after the birth. There are severely lower numbers of neurons in regions of the hippocampus and cerebellum of Purα-/- mice versus those of age-matched Pur+/+ littermates, suggesting that Purα plays a critical role in neurogenesis [19]. Immunohistochemical analysis of the MCF7 marker for DNA replication reveals a lack of proliferation in precursor cells in this region, implying that Purα developmentally regulates DNA replication in specific cell types in the brain. Primary cultures of mouse embryo fibroblasts (MEFs) derived from the Purα-/- and Purα+/+ control mice have been used as a useful model to test the role of Purα in DNA repair and checkpoints control [21,22]. This in vitro model system provided a valuable approach in extending the mechanistic studies to various relevant processes such as DNA repair and cell cycle control although the adult Purα knockout mice could not be generated.
The research work in Wang H’s group indicated that Purα-/- cells are more sensitive to HU and CPT than wild type cells, and that expression of wild type Purα abrogated this hyper-sensitivity [21,22]. It also very interesting that HU triggers translocation of Purα to the nucleus, where it co-localize with Rad51 and PARP. Furthermore, caffeine, an inhibitor of ATM and ATR, diminished the nuclear translocation. Therefore, it is likely that Purα impacts on DNA repair and checkpoint regulation to maintain genomic stability.
Purα is essential for the control of cell growth, presumably due to its ability to interact with several cell cycle controls, as Purα null cells exhibit a fast growth rate and display characteristics of immortalized cells. Re-transfection of Purα gene back into Purα knockout cells represses the observed phenotypes and restores the normal cell growth, perhaps due to its ability to associate with and modulate the function of various cell cycle regulators such as pRb, E2f1, Cyclin A, Cyclin B and Cdk5. Curiously, chromosomal abnormalities were detected in Purα null cells upon treatment with genotoxic agents. As DNA damage has been implicated in neurodegenerative disorders, one may envision a role of Purα, either dependent or independent from cell cycle, in the maintenance of DNA integrity in neuronal cells. It has been reported that Purα null cells are significantly more sensitive to various DNA damaging agents than their wild type counterparts, and that re-transfection the Purα back into Purα-/- MEFs can reverse the phenotype. Furthermore, tagging of Purα with GFP show that HU treatment triggers translocation of Purα to the nucleus. Interestingly, caffeine, an inhibitor of ATM and a wellknown abrogater of DNA-damage-induced checkpoint responses, inhibits the nuclear translocation of Purα. Irradiated Pur-/- MEF cells fail to arrest in G1 but show a strong G2 checkpoint than that in Purα+/+ MEF cells, suggesting that Purα regulates the G1 to S transition. Interestingly, treatment with hydroxyurea induced poly (ADP-ribose) modification of a higher molecular weight protein other than poly (ADP-ribose) polymerase 1 (PARP-1) in Purα-/- MEF cells, but not in Purα+/+ MEF cells. A GST pull-down assay shows that Purα interacts with PARP-1. These observations led to the hypothesis that Purα is an important component of DNA repair and cell cycle checkpoint whose activity is critical for the maintenance of genomic integrity in neuronal cells [86].
The effects of Purα on DNA repair have been studied in our laboratory recently and we constructed the lentivirus expression plasmid with Purα RNAi and overexpression constructs [87]. We investigated the protective effects of Purα protein on rat hippocampus DNA damage induced by epilepsy and the effects of Purα protein on DNA damage and repair have also been investigated. The high tittered lentivirion of Purα overexpression, RNAi as well as lentivirus empty vector as control were separately injected into rat hippocampus guided by stereotaxic apparatus. The level of Purα expression and knock down were checked in the 14th days after the virion injection with fluorescent slides and western blotting to confirm that virus has already infected the hippocampus tissues. Pilocarpine was used to induce epilepsy by abdominal injection. The experimental animals were executed 1 hours after the epileptic onset and the hippocampus samples were collected for immunohistochemical staining and western blotting assay to examine the pertinent protein expression to investigate the protective effects of Purα on DNA damage and repair. The results demonstrated that pilocarpine can induce epileptic onset, immunohistochemistry exhibited that γH2AX, a landmark protein for DNA damage, has higher content in CA1 region of rat hippocampus. The damage became aggravated when Purα protein has been knocked down, but in Purα overexpression group, the damage became alleviated obviously. The results of western blotting illustrated that the proteins associated with DNA damage such as Parp-1, Ku80, XRCC4 has higher expression level when Purα was knocked down, but on the other hand, these proteins have lower expression when Purα was overexpressed. All these results indicate that Purα protein can protect the DNA damage caused by epilepsy and also participated in the repair process of DNA damage [88].
Conclusion
DNA damage is an inevitable processes in our body since the genomic material is under a constant attack from numerous exogenous and endogenous agents. To response to DNA damage, a serious DNA damage repair pathway will be activated and the damaged DNA will be repaired through the different mechanisms. The damaged DNA is not repaired or the ability of DNA repair is deficiency, the stability of the genomic DNA will be disturbed and the post-mitotic neuron will be provoked to reentry into cell cycle. Since lack of the necessary cell cycle proteins, the reentry into the cell cycle for the post-mitotic neuron will be lethal and the fate of the reentry will be a disastrous event and the cells will undergo death, apoptosis or mutation, in this way neurodegeneration will be caused. Purα is a transcriptional factors that can bind to DNA and RNA in a special way and involves in the cell growth and proliferation, initiation of replication, transcription, translation and RNA transportation. Purα is also an important factors for maintenance of stability and integrity of genomic DNA in nervous system. Purα knock out mice have been genetically engineered and lack of Purα exhibited a severe defects in brain development. Purα-/- MEFs exhibited immortalized growth phenotype and prone to genotoxic agents such as HU, CPT as well as the ion radiation. Re-transfected Purα back into Purα-/- MEFs could reverse these observed phenomenon. Purα also protects the cells from genotoxic agent induced DNA damage as well as the epilepsy induced DNA damage in rat hippocampus.
Acknowledgments
This work was supported by a Chinese National scientific foundation grant to Dr. Jianqi Cui (81260197) and The Chinese National 973 project grants to Dr. Tao Sun (2012CB722408) and Dr. Jianguo Niu (2014CB560711).
6739
References
- Madabhushi R, Pan L, Tsai LH (2014) DNA damage and its links to neurodegeneration. Neuron 83: 266-282.
- Rulten SL, Caldecott KW (2013) DNA strand break repair and neurodegeneration. DNA Repair (Amst) 12: 558-567.
- Jeppesen DK, Bohr VA, Stevnsner T (2011) DNA repair deficiency in neurodegeneration. ProgNeurobiol 94: 166-200.
- Calero M, Gómez-Ramos A, Calero O, Soriano E, Avila J, et al. (2015) Additional mechanisms conferring genetic susceptibility to Alzheimer's disease. Front Cell Neurosci 9: 138.
- Morishima-Kawashima M (2014) Molecular mechanism of the intramembrane cleavage of the β-carboxyl terminal fragment of amyloid precursor protein by β-secretase. Front Physiol 5: 463.
- Stancu IC, Vasconcelos B, Terwel D, DewachterI (2014) Models of β-amyloid induced Tau-pathology: the long and "folded" road to understand the mechanism. MolNeurodegener 9: 51.
- Serý O, Povová J, Míšek I, Pešák L, Janout V (2013) Molecular mechanisms of neuropathological changes in Alzheimer's disease: a review. Folia Neuropathol 51: 1-9.
- Iyama T, Wilson DM 3rd (2013) DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12: 620-636.
- Neema M, Navarro-Quiroga I, Chechlacz M, Gilliams-Francis K, Liu J, et al. (2005) DNA damage and nonhomologous end joining in excitotoxicity: neuroprotective role of DNA-PKcs in kainic acid-induced seizures. Hippocampus 15: 1057-1071.
- Kwiatkowski D, Czarny P, Galecki P, Bachurska A, Talarowska M, et al. (2015) Variants of Base Excision Repair Genes MUTYH , PARP and XRCC in Alzheimer's Disease Risk. Neuropsychobiology 71: 176-186.
- Crespan E, Hübscher U, Maga G (2015) Expansion of CAG triplet repeats by human DNA polymerases λ and β in vitro, is regulated by flap endonuclease and DNA ligase 1. DNA Repair (Amst) 29: 101-111.
- Ho SR, Mahanic CS, Lee YJ, Lin WC (2014) RNF144A, an E3 ubiquitin ligase for DNA-PKcs, promotes apoptosis during DNA damage. ProcNatlAcadSci U S A 111: E2646-2655.
- Reiling E, Dollé ME, Youssef SA, Lee M, Nagarajah B, et al. (2014) Theprogeroid phenotype of Ku80 deficiency is dominant over DNA-PKCS deficiency. PLoS One 9: e93568.
- Kim KB, Kim DW, Park JW, JeonYJ, Kim D, et al. (2014) Inhibition of Ku70 acetylation by INHAT subunit SET/TAF-Iβ regulates Ku70-mediated DNA damage response. Cell Mol Life Sci 71: 2731-2745.
- Maréchal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring HarbPerspectBiol 5.
- Cooper TJ, Wardell K, Garcia V, Neale MJ (2014) Homeostatic regulation of meiotic DSB formation by ATM/ATR. Exp Cell Res 329: 124-131.
- Yan S, Sorrell M, Berman Z (2014) Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci 71: 3951-3967.
- Johnson EM (2003) The Pur protein family: clues to function from recent studies on cancer and AIDS. Anticancer Res 23: 2093-2100.
- Khalili K, Del Valle L, Muralidharan V, GaultWJ, Darbinian N, et al. (2003) Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol 23: 6857-6875.
- Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, et al. (2012) Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum Mol Genet 21: 473-484.
- Wang H, Wang M, Reiss K, Darbinian-Sarkissian N, Johnson EM, et al. (2007) Evidence for the involvement of Puralpha in response to DNA replication stress. Cancer BiolTher 6: 596-602.
- Wang H, White MK, Kaminski R, Darbinian N, Amini S, et al. (2008) Role of Puralpha in the modulation of homologous recombination-directed DNA repair by HIV- Tat. Anticancer Res 28: 1441-1447.
- Lalani SR, Zhang J, SchaafCP, Brown CW, Magoulas P, et al. (2014) Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3microdeletion syndrome. Am J Hum Genet 95: 579-583.
- Hunt D, Leventer RJ, Simons C, Taft R, Swoboda KJ, et al. (2014) Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet 51: 806-813.
- Fasullo M, Endres L (2015) Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int J MolSci 16: 9431-9449.
- ChrzanowskaKH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M (2012) Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis 7: 13.
- Lee J, Hwang YJ, Ryu H, Kowall NW, Ryu H (2014) Nucleolar dysfunction in Huntington's disease. BiochimBiophysActa 1842: 785-790.
- Fernández-Moriano C, González-Burgos E, Gómez-SerranillosMP (2015) Mitochondria-Targeted Protective Compounds in Parkinson's and Alzheimer's Diseases. Oxid Med Cell Longev 2015: 408927.
- AbnerCW, McKinnon PJ (2004) The DNA double-strand break response in the nervous system. DNA Repair (Amst) 3: 1141-1147.
- Caldecott KW (2004) DNA single-strand breaks and neurodegeneration. DNA Repair (Amst) 3: 875-882.
- Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, et al. (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41: 549-561.
- Hamdane M, Delobel P, Sambo AV, Smet C, Bégard S, et al. (2003) Neurofibrillary degeneration of the Alzheimer-type: an alternate pathway to neuronal apoptosis? BiochemPharmacol 66: 1619-1625.
- Kim D, Tsai LH (2009) Linking cell cycle reentry and DNA damage in neurodegeneration. Ann N Y AcadSci 1170: 674-679.
- Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, et al. (2001) Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 24: 25-31.
- Liu DX, Greene LA (2001) Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 32: 425-438.
- Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, et al. (1998) Response of postmitotic neurons to X-irradiation: implications for the role of DNA damage in neuronal apoptosis. J Neurosci 18: 147-155.
- Morris EJ, Geller HM (1996) Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol 134: 757-770.
- Jacobsen E, Beach T, Shen Y, Li R, Chang Y (2004) Deficiency of the Mre1 DNA repair complex in Alzheimer's disease brains. Brain Res Mol Brain Res 128: 1-7.
- Nouspikel T, Hanawalt PC (2003) When parsimony backfires: neglecting DNA repair may doom neurons in Alzheimer's disease. Bioessays 25: 168-173.
- Lu T, Pan Y, Kao SY, Li C, Kohane I, et al. (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429: 883-891.
- Prakash S, Prakash L (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev 16: 1872-1883.
- Haber JE (1999) DNA recombination: the replication connection. Trends BiochemSci 24: 271-275.
- Iliakis G, Pantelias G, Kurtzman S (1991) Mechanism of radiosensitization by halogenated pyrimidines: effect of BrdU on cell killing and interphase chromosome breakage in radiation-sensitive cells. Radiat Res 125: 56-64.
- IliakisG (1991) The role of DNA double strand breaks in ionizing radiation-induced killing of eukaryotic cells. Bioessays 13: 641-648.
- KowalczykowskiSC (2000) Initiation of genetic recombination and recombination-dependent replication. Trends BiochemSci 25: 156-165.
- Rothstein R, Michel B, Gangloff S (2000) Replication fork pausing and recombination or "gimme a break". Genes Dev 14: 1-10.
- Baumann P, West SC (1998) Role of the human RAD5 protein in homologous recombination and double-stranded-break repair. Trends BiochemSci 23: 247-251.
- Critchlow SE, Jackson SP (1998) DNA end-joining: from yeast to man. Trends BiochemSci 23: 394-398.
- PetriniJH, Bressan DA, Yao MS (1997) The RAD52 epistasis group in mammalian double strand break repair. SeminImmunol 9: 181-188.
- LieberMR (1999) The biochemistry and biological significance of nonhomologous DNA end joining: an essential repair process in multicellular eukaryotes. Genes Cells 4: 77-85.
- Riballo E, Critchlow SE, TeoSH, Doherty AJ, Priestley A, et al. (1999) Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. CurrBiol 9: 699-702.
- Wang H, Perrault AR, Takeda Y, Qin W, Wang H, et al. (2003) Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res 31: 5377-5388.
- Moynahan ME, Chiu JW, KollerBH, Jasin M (1999) Brca controls homology-directed DNA repair. Mol Cell 4: 511-518.
- Wang H, ZengZC, Bui TA, DiBiaseSJ, Qin W, et al. (2001) Nonhomologous end-joining of ionizing radiation-induced DNA double-stranded breaks in human tumor cells deficient in BRCA or BRCA2. Cancer Res 61: 270-277.
- Xia F, Taghian DG, DeFrankJS, ZengZC, Willers H, et al. (2001) Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. ProcNatlAcadSci U S A 98: 8644-8649.
- Couedel C, Mills KD, Barchi M (2004) Collaboration of homologous recombination and nonhomologous end-joining factors for the survival and integrity of mice and cells. Genes Dev 18: 1293-1304.
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T (1998) Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. CurrBiol 8: 1395-1398.
- Deans B, Griffin CS, Maconochie M, Thacker J (2000) Xrcc2 is required for genetic stability, embryonic neurogenesis and viability in mice. EMBO J 19: 6675-6685.
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, et al. (1998) A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95: 891-902.
- Gu Y, Sekiguchi J, Gao Y, Dikkes P, Frank K, et al. (2000) Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. ProcNatlAcadSci U S A 97: 2668-2673.
- Frank KM, Sharpless NE, Gao Y, SekiguchiJM, Ferguson DO, et al. (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5: 993-1002.
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, et al. (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404: 897-900.
- Lee Y, Barnes DE, Lindahl T, McKinnon PJ (2000) Defective neurogenesis resulting from DNA ligase IV deficiency requires Atm. Genes Dev 14: 2576-2580.
- Sekiguchi J, Ferguson DO, Chen HT, Yang EM, Earle J, et al. (2001) Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. ProcNatlAcadSci U S A 98: 3243-3248.
- Caldecott KW (2003) DNA single-strand break repair and spinocerebellar ataxia. Cell 112: 7-10.
- Haas S, Gordon J, Khalili K (1993) A developmentally regulated DNA-binding protein from mouse brain stimulates myelin basic protein gene expression. Mol Cell Biol 13: 3103-3112.
- Haas S, Thatikunta P, Steplewski A, Johnson EM, Khalili K, et al. (1995) A 39-kD DNA-binding protein from mouse brain stimulates transcription of myelin basic protein gene in oligodendrocytic cells. J Cell Biol 130: 1171-1179.
- Bergemann AD, Ma ZW, Johnson EM (1992) Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein. Mol Cell Biol 12: 5673-5682.
- Bergemann AD, Johnson EM (1992) The HeLaPur factor binds single-stranded DNA at a specific element conserved in gene flanking regions and origins of DNA replication. Mol Cell Biol 12: 1257-1265.
- Ma ZW, Bergemann AD, Johnson EM (1994) Conservation in human and mouse Pur alpha of a motif common to several proteins involved in initiation of DNA replication. Gene 149: 311-314.
- Gallia GL, Johnson EM, Khalili K (2000) Puralpha: a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res 28: 3197-3205.
- Johnson EM, Daniel DC, Gordon J (2013) Thepur protein family: genetic and structural features in development and disease. J Cell Physiol 228: 930-937.
- White MK, Johnson EM, Khalili K (2009) Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle 8: 1-7.
- Darbinian N, Gallia GL, Khalili K (2001) Helix-destabilizing properties of the human single-stranded DNA- and RNA-binding protein Puralpha. J Cell Biochem 80: 589-595.
- Johnson EM, Chen PL, KrachmarovCP, Barr SM, Kanovsky M, et al. (1995) Association of human Pur alpha with the retinoblastoma protein, Rb, regulates binding to the single-stranded DNA Pur alpha recognition element. J BiolChem 270: 24352-24360.
- Darbinian N, Gallia GL, Kundu M, Shcherbik N, Tretiakova A, et al. (1999) Association of Pur alpha and E2F- suppresses transcriptional activity of E2F-1. Oncogene 18: 6398-6402.
- Tretiakova A, Steplewski A, Johnson EM, Khalili K, Amini S (1999) Regulation of myelin basic protein gene transcription by Sp andPuralpha: evidence for association of Sp and Puralpha in brain. J Cell Physiol 181: 160-168.
- Safak M, Gallia GL, Khalili K (1999) Reciprocal interaction between two cellular proteins, Puralpha and YB-, modulates transcriptional activity of JCVCY in glial cells. Mol Cell Biol 19: 2712-2723.
- Mishra M, Del Valle L, Otte J, Darbinian N, Gordon J (2013) Pur-alpha regulates RhoA developmental expression and downstream signaling. J Cell Physiol 228: 65-72.
- Gallia GL, Safak M, Khalili K (1998) Interaction of the single-stranded DNA-binding protein Puralpha with the human polyomavirusJC virus early protein T-antigen. J BiolChem 273: 32662-32669.
- Itoh H, WortmanMJ, Kanovsky M, Uson RR, Gordon RE, et al. (1998) Alterations in Pur(alpha) levels and intracellular localization in the CV- cell cycle. Cell Growth Differ 9: 651-665.
- Stacey DW, Hitomi M, Kanovsky M, Gan L, Johnson EM (1999) Cell cycle arrest and morphological alterations following microinjection of NIH3T3 cells with Pur alpha. Oncogene 18: 4254-4261.
- Barr SM, Johnson EM (2001) Ras-induced colony formation and anchorage-independent growth inhibited by elevated expression of Puralpha in NIH3T3 cells. J Cell Biochem 81: 621-638.
- Darbinian N, Gallia GL, King J, Del Valle L, Johnson EM, et al. (2001) Growth inhibition of glioblastoma cells by human Pur(alpha). J Cell Physiol 189: 334-340.
- Yongling L, Juan C, Zhongfa J (2015) The Regulatory effects of Pura on PARP gene expression and DNA repair. Chinese Journal of Cell Biology.
- Zhongfa J, Lin M, Zhengquan H (2014) The construction and application of Lentivirus vectors of Overxpression and RNAi for Pura gene. Journal of Ningxia Medical University 36: 620-634.
- Qiang L, Tao S, Zheng Y (2015) The protective effects of Pura on rat hippocampus DNA damage induced by epilepsy. Chinese Journal of Medicine.


