Keywords
Cardiovascular disease, circulatory system, smoking, tobacco
Introduction
Smoking is a major risk factor for cardiovascular morbidity and mortality, and is considered to be the leading preventable cause of death in the world. [1,2] Internationally, 25% of middle-aged cardiovascular deaths are attributable to smoking. [3] The European Society of Cardiology reported recently that smoking causes 28% of cardiovascular deaths in men aged 35 to 69 years and 13% in women of the same age. [4] In the European Region of the World Health Organisation (WHO), smoking is the second most important risk factor in the burden of disability-adjusted life years and is the primary risk factor for premature mortality, associated with about 1.6 million deaths each year. [5] In the European Union (EU), 15% of all-cause deaths are attributed to smoking, amounting to 655,000 smoking-related deaths each year, [6] while in Greece, the percentage of deaths from any smoking-associated cause, among individuals ages 35 and older, has been estimated to be 18.1%. [7] Based on estimates by the WHO, tobacco continues to kill nearly 6 million people each year—including more than 600,000 passive smokers—through heart disease, lung cancer, and other illnesses; [1] that is one and a half million more than the corresponding estimate for 1990. [3] If current trends continue, the death toll is projected to reach more than 8 million per year by 2030. [1]
Smoking ranks among the top causes of cardiovascular disease, including coronary heart disease, ischemic stroke, peripheral artery disease and abdominal aortic aneurysm. [4] It is also associated with an increased risk of certain types of cancer, and is a major cause of chronic obstructive pulmonary disease. [1,2] Smoking, either active or passive, can cause cardiovascular disease via a series of interdependent processes, such as enhanced oxidative stress, haemodynamic and autonomic alterations, endothelial dysfunction, thrombosis, inflammation, hyperlipidaemia, or other effects. [8] Even exposure to small quantities—e.g. occasional smoking, passive smoking, a few cigarettes per day—is sufficient to have deleterious consequences. [9] Cigarette smoke contains more than 4000 chemical substances that have harmful effects on cardiovascular function. [10] These include nicotine, carbon monoxide (CO), oxidative gases, polycyclic aromatic hydrocarbons, carbonyls, butadiene, minerals, carbon disulphide, and benzene. Although many of the toxic substances contained in tobacco smoke are generic products of the combustion of organic materials, exposure to smoking involves contact with two substances that are specific to tobacco smoke and are known to be damaging to the health: nicotine and CO. [11]
Despite all the publicity concerning the documented adverse effects of smoking on public health, smoking prevalence still remains high in the EU, where approximately 30% of citizens are smokers. [12] Greece suffers from an enormous smoking-related public health problem, having the highest proportion of smokers in the EU (42%). [12] According to WHO estimates, in Greece 63.4% of adult males and 39% of females ≥15 years are smokers, [13] while in young adults, aged 19-30 years, the smoking prevalence is 37.6%. [14] The purpose of this article is to provide a brief description of the effects of smoking, and in particular the effects of nicotine and CO, on cardiovascular function, providing essential information that could contribute to reducing the smoking epidemic and its consequences for cardiovascular health.
Nicotine
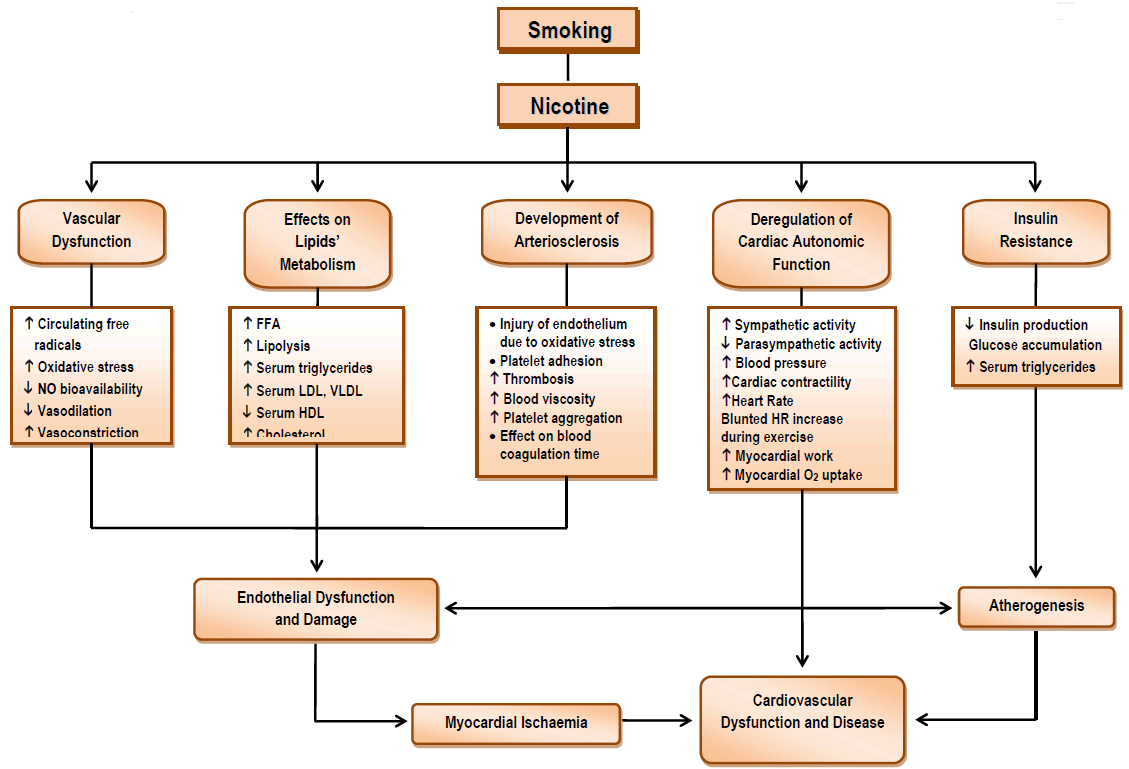
Nicotine is classed as an alkaloid (like morphine and cocaine) and meets the criteria of a highly addictive drug. One cigarette delivers 1.2-2.9 mg of nicotine, and the typical one pack-per-day smoker absorbs 20-40 mg of nicotine each day. [15] As an addictive drug, nicotine has 2 very potent effects: it is a stimulant and it is also a depressant. [16] Nicotine deregulates cardiac autonomic function, boosts sympathetic activation, raises heart rate, causes coronary and peripheral vasoconstriction, increases myocardial workload, and stimulates adrenal and neuronal catecholamine release. [17] In addition, nicotine is associated with insulin resistance, increased serum lipid levels, and intravascular inflammation that contributes to the development of atherosclerosis (Figure 1). [17]
Vascular Function
There are ample published data documenting that chronic exposure to tobacco smoke leads to a pathological alteration of endothelial function. Endothelial dysfunction may be caused by metabolic (dyslipidaemia), environmental (smoking), and physical (arterial hypertension) factors, or by inflammation that provokes pathological conditions. [18] It is characterised by an imbalance between vasodilatory and vasoconstrictive substances originating from the endothelium, anticoagulant and procoagulant mechanisms, growth factors and growth inhibitors. [18]
Under normal conditions, the free radicals circulating in the human body are neutralised by defensive mechanisms. However, if their concentrations within the blood should increase greatly because of excessive exposure to harmful factors such as smoking, then they cannot be regulated and may cause dangerous mutations that destroy cells. In these circumstances, oxidative stress is seen to arise. [10] The term “oxidative stress” refers to the total of the intracellular and extracellular conditions that lead to chemical or metabolic production of reactive oxygen species (ROS). [11] Smoke exists mainly in two states: the gaseous (which includes CO) and the solid (tar). In both these states, it contains a large quantity of free radicals. [8] Pryor and Stone determined that 1 g of tar from cigarette smoke contains more than 1017 long-lived free radicals (hours to months), while 1 g volatile fraction of smoke contains 1015 short-lived free radicals (seconds). [19] Chronic exposure to tobacco also weakens the antioxidant defensive mechanisms that regulate these large numbers of smoking-induced free radicals, leading to a significant increase in oxidative stress. [8] Oxidative stress, the oxidation of lipids, proteins, and DNA, is directly associated with atherogenesis. [10] An indicative finding is that when levels of isoprostanes (indexes of lipid peroxidation and oxidative damage) were measured in smokers, their levels were found to be higher than in non-smokers. [11] The reaction of nitric oxide (NO) with the free radicals contained in smoke reduces NO’s bioavailability, interfering with its vasodilatory, antithrombotic, anti-inflammatory, and antioxidant effects, as well as its influence on endothelium permeability and myocardial function (reducing the diastolic distensibility of the left ventricle). [20] The alteration in biosynthesis of NO and its decreased activity, [21] in combination with the smoking-induced reduction in prostacyclin production [22] and the direct toxic effect of nicotine on endothelial cells that causes direct structural damage, [17] are important factors that may lead to endothelial dysfunction (Figure 1).
Using an extract of cigarette tobacco or its isolated ingredients, such as nicotine, many in vitro studies have found that smoking is associated with reduced NO availability. It has been shown that nicotine concentration in smokers’ blood serum reduces the availability of NO in human umbilical vein endothelial cells (HUVECs), as well as in human coronary artery endothelial cells, leading to a reduction in the brachial artery’s endothelium-dependent vasodilation. [8] Using this model, Barua et al. demonstrated that exposure to smokers’ sera decreased NO availability in both HUVECs and human coronary artery endothelial cells, by altering the expression and activity of the endothelial NO synthase enzyme. [21] In addition, they noted a significant correlation between flow-mediated brachial artery endothelium-dependent vasodilation and NO bioavailability from cultured HUVECs exposed to serum from the same individuals. On the other hand, CO, which is significantly elevated in smokers, inhibits the creation of NO and takes its place in haemoglobin bonds. [23] These findings lead to the conclusion that the large quantities of free radicals contained in smoke enhance oxidative stress and, in combination with reduced NO bioavailability, nicotine-induced vasoconstriction and impaired vasodilation, may lead to endothelial dysfunction. The consequent endothelial damage contributes to the formation and progression of atheromatous plaque, and reduces blood flow via thrombosis and vasospasm, thus causing cardiovascular disease. [18,24]
Lipid Metabolism
Tobacco smoke, and specifically nicotine, has a significant effect on lipid metabolism and the regulation of lipid levels in the blood. [25] Therefore, cigarette smoke could promote atherosclerosis, in part, via its effects on the lipid profile. [8] It has been shown that, in the presence of already increased serum lipid levels, smoking exacerbates the condition. [26]
Smoking is associated with significantly elevated serum concentrations of total cholesterol and triglycerides. [25] In addition, several studies have shown a tendency for low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL) cholesterol to be slightly higher in smokers. [27] These associations seem to be dose dependent. [25] On the other hand, smoking lowers serum concentrations of high-density lipoprotein (HDL) cholesterol, a powerful protective factor against the development of atherosclerosis. [28,29] The difference is usually small, 5 mg/dl or less, but this difference represents a 10% decrease and would be expected to affect atherogenesis to a significant degree. [27] Giving up smoking improves HDL levels, regardless of body weight, contributing to an improvement in cardiovascular health after smoking cessation. [30]
It is possible that oxidative damage to protein and lipid constituents may explain the way in which cigarette smoke affects plasma LDL and HDL. [10] Cigarette smoking increases the oxidative modification of LDL. [8] Exposure to cigarette smoke extract also decreases the plasma activity of paraoxonase, an enzyme that protects against LDL oxidation. [8] There are two potential mechanisms by which reactive smoke components can produce their deleterious effects on essential plasma constituents: 1) indirectly, gas-phase cigarette smoke may activate macrophages and neutrophils in the lung, which may release enzymes and oxidants capable of damaging lipids and proteins; 2) directly, since the lung possesses an extremely large surface area for gas exchange, it is possible that gas-phase cigarette smoke components interact with plasma constituents in the interstitial fluid. [29]
Additional mechanisms have been proposed to explain the link between smoking and changes in serum lipid and lipoprotein concentrations. Nicotine stimulates the release of adrenaline by the adrenal cortex, leading to the increased serum concentrations of free fatty acids (FFA) observed in smokers. As a result, lipolysis is increased along with the blood’s triglyceride capacity. FFA are known to stimulate the hepatic secretion of VLDL and hence triglycerides. [25,26] The increased release of FFA in the heart raises myocardial oxygen consumption, adding to the myocardial workload. [31-33] A complementary finding is that FFA also stimulate the hepatic synthesis and secretion of cholesterol. [25] The smoking-induced changes in lipid metabolism, the increased LDL/VLDL and decreased HDL levels, in combination with the destruction of vascular endothelium, are associated with a greater incidence of atherosclerosis in smokers. Thus, hypercholesterolaemia and smoking are among the most important factors that may lead to coronary artery disease. [34,35]
Arteriosclerosis
Arteriosclerosis is a general term that includes almost all the arterial disorders that lead to the thickening and hardening of all kinds of artery. Atherosclerosis is a specific form of arteriosclerosis, whose most characteristic feature is the concentration of lipids in the intima of large elastic arteries (aorta) and medium-sized muscular arteries (coronary, femoral, carotid, etc.). [36]
Smoking is considered to be a significant risk factor for the development of atherosclerosis. The atherosclerotic effects of cigarette smoke are due to a substantial degree to thrombosis-related events. [11] The accumulation of platelets coating the arterial wall at sites where there is turbulent
blood flow or endothelial injury may be the prodromal stage for the creation of atheromatous plaque. [37] Nicotine is considered to be responsible for an increase in blood viscosity and platelet aggregation, since it prevents the production of prostacyclins that would limit platelet aggregation. [26] Platelet adhesion increases the production of thrombi, splits the coronary artery intima, accelerates the process of atheromatous plaque creation, and is associated with an increased risk of cardiac ischaemia. [11] In addition, nicotine affects prostaglandin metabolism, weakening the vessel’s defence against platelet deposition. [38] The increase in platelet aggregation, the effect of nicotine on blood coagulation time, and the increase in blood viscosity, in combination with the increase in levels of LDL and VLDL, the reduction in HDL, and inflammatory processes, promote the creation of atheromatous plaque and the development of atherosclerosis (Figure 1). [16,17,27] It is thus likely that chronic smoking, by increasing peripheral vascular resistances in this way, may lead to an increase in cardiac afterload and a consequent reduction in stroke volume. [39]
Circulatory levels of fibrinogen, one of the most powerful predictive markers of coronary events, are elevated in smokers. The increase in fibrinogen levels acts in combination with the increase in red cell mass from long-term exposure to CO, increasing blood viscosity and boosting the activation of platelets, hence increasing the risk of atherogenesis. Increased fibrinogen levels in the blood circulation can also lead to the development of atherosclerosis, with a direct effect on the increase in platelet aggregation. [40]
Tissue factor (TF)—otherwise known as tissue platelet factor, or factor III, or thrombokinase, or CD 142—is a protein found in endothelial tissue, platelets, and leucocytes, and is essential for the initiation of thrombus formation by zymogen prothrombin. [41] TF is expressed by cells that are normally not exposed to blood flow, such as sub-endothelial cells (e.g. smooth-muscle cells) and the cells that surround blood vessels (e.g. fibroblasts). [41] This can change when blood vessels are damaged—for example by physical injury, or rupture, or atherosclerotic plaque. TF is present in atherosclerotic plaque and can promote thrombogenesis and possibly propagation of the thrombus to the already existing atherosclerosis. Sambola et al. found that smoking increased plasma TF levels in smokers who smoked 10 or more cigarettes per day, with a smoking history of 10 or more years. [42]
Atherogenesis and coronary artery disease are the result of inflammatory processes. The fact that smoking is associated with inflammation implies that inflammation may be one of the mechanisms via which cigarette smoking leads to cardiovascular dysfunction. C-reactive protein (CRP) and levels of white blood cells are markers of inflammation, and are thus associated with atherosclerosis and an increased risk of cardiovascular disease. [43] Levels of CRP and white blood cells appear to be higher in smokers than in non-smokers. Furthermore, there appears to be a relation between the extent of smoking and the white blood cell count. [44] Dietrich et al. claimed that the increase in CRP observed in smokers is proportional to both the quantity and the years of smoking. [44]
Overall, nicotine boosts sympathetic activity, stimulates the release of neurotransmitters, causes coronary and peripheral vasoconstriction, and elevates blood pressure. Furthermore, nicotine increases lipolysis, leads to increased levels of free fatty acids, increases oxidative stress, endothelial damage and dysfunction, and promotes vessel inflammation, contributing significantly to the development of atherosclerosis and cardiovascular disease.
Autonomic Nervous System
There is an established link between abnormal heart rate (HR) responses at rest and during exercise, autonomic dysfunction and cardiovascular health. [45,46] On the other hand, chronic smoking is associated with dysfunction of the autonomic nervous system, [47-49] and the abnormal HR responses to tobacco may be implicated in the link between smoking and cardiovascular disease. [50-53] Although the precise mechanism of action of smoke ingredients is still under investigation, all proposed hypotheses state that the main effects of smoking on cardiovascular function are associated with the direct or indirect action of nicotine on the neuroregulation of the circulatory system, wherein sympathetic activity is increased and parasympathetic activity is reduced (Figure 1). [36] The nicotine-induced sympathetic overdrive causes the adrenal medulla to increase the secretion of both epinephrine and norepinephrine into the circulating blood. [54] In addition, nicotine stimulates the vasomotor centre of the medulla, causing secretion of norepinephrine from local deposits. Subsequently, secretion of catecholamines from the free nerve endings of the sympathetic nerves and the local release of epinephrine and norepinephrine are increased. In addition, vasoconstriction of coronary vessels occurs, the biosynthetic capacity of prostacyclin is reduced, and endothelial function is impaired. [55,56] The stimulation of catecholamine secretion, in combination with the depressed production of prostacyclins (potent vasodilators), results in an acute rise in blood pressure, a significant rise in HR, an increase in cardiac contractility, and a significant increase in myocardial work. [36] Nicotine affects cardiovascular function both directly, as described previously, and indirectly, through a series of neurohormonal changes. In particular, nicotine molecules interact with and activate the brain's acetylcholine receptors (nAChRs), whose prolonged activation may desensitize a proportion of them. [57] The activation of nAChRs by nicotine boosts the release of neurotransmitters, while altering the function of some of them—such as norepinephrine, dopamine, serotonin (5-HT), and endogenous opioid peptides—thus modifying the action of the peripheral nervous system and causing cigarette addiction. [58]
Heart Rate at Rest
Smoking has been associated with increased resting HR values in healthy adults, regardless of age or sex. [59-61] Minami et al. found that the HR is on average 7 bpm faster in smokers than in non-smokers; [62] this finding is in line with data reported by Papathanasiou et al., indicating that smokers had a significantly higher resting HR compared with non-smokers in both female (76.4 bpm vs. 70.0 bpm, p=0.001) and male (72.8 vs. 66.3, p=0.004) subjects. [63] In particular, smoking is associated with selective alterations in cardiac autonomic control. [64,65] More specifically, smoking, acting at peripheral sympathetic sites, increases circulating levels of catecholamines,66 augments sympathetic outflow, [49,67] and causes a long-term reduction in vagal drive.65 This sympathetic predominance, seen even in young heavy smokers, [65] is also associated with impaired baroreflex function, [49,64] leading to a marked increase in HR at rest.
Heart Rate during Exercise
During exercise, the increased metabolic demands are met by an increased cardiac output, achieved through an augmentation in HR and stroke volume. The elevation of HR, which is associated with age, HRrest and exercise capacity, [68,69] is regulated by exercise-induced autonomic control, where sympathetic activity increases and vagal tone is reduced. The HR elevation peaks at maximal exercise, when healthy subjects achieve an actual HRmax close (±10 bpm) to their age-predicted HRmax. [69] An impaired HR response to exercise and failure to reach >80% of the age-predicted HRmax, known as chronotropic incompetence, [69] are associated with autonomic imbalance and are important prognostic markers of cardiovascular health. [50,52] In many HR-related studies in healthy adults, smoking was found to blunt HR elevation during progressive exercise and to lower the maximum HR achieved, [50,63,70] posing an increased risk to smokers’ health. [48,53] Adaptations to chronic smoking, such as down-regulation of β-adrenergic receptors, have been used to explain smokers’ blunted HR response to exercise. [48,71] Long-term smoking has been found to decrease the density of lymphocyte or adipose tissue β-receptors, down-regulating the β-receptors of the cardiovascular system. [66]
Heart Rate Recovery
On the completion of vigorous exercise, sympathetic activity withdraws and vagal reactivation mediates the rate at which HR decreases, making the post-exercise HR decline an important reference marker for cardiac autonomic control. [72,73] Attenuated HR decline during recovery is an important surrogate for underlying autonomic dysfunction that is associated with increased cardiovascular morbidity and mortality. [51,52,74] In many epidemiological HR-related studies in healthy middle-aged populations, smoking was inversely associated with HR decline during recovery. [51,74] Other studies, though few in number, have shown that an attenuated HR decline was also present in young smokers after submaximal [61] or maximal exercise. [63]
Smoking and Insulin Resistance
Insulin has an effect on practically all the tissues of the body, either directly or indirectly, and is characterised as a storage hormone because of its anabolic action on all three main dietary groups: namely, carbohydrates, fats, and proteins. Insulin is associated with specific receptors in the cellular membrane. The basic functions of the insulin receptor are to recognise and to bind the hormone with the target cell, and to transmit its metabolic action. If one of these functions is disturbed, insulin resistance is manifested. [75] Insulin resistance, metabolic syndrome, and glucose intolerance are regarded as disturbances with a common background and strong interrelations. [76]
Nicotine is known to increase sympathetic activity, to raise circulating levels of catecholamines, growth hormone, adrenocorticotropic hormone, cortisol, prolactin, and beta-endorphin, and to decrease oestrogen levels; [77] all these effects are strongly antagonistic to insulin’s action. Thus, smoking reduces insulin production, slowing glucose catabolism and leading to its accumulation in the body. Nicotine may also increase insulin resistance directly. It has been shown that the increase in insulin resistance was halted after nicotine replacement was stopped, and even improved during continuous weight gain, [78] implying that nicotine rather than weight gain may be responsible for the initial increase in insulin resistance observed in some smoking-related studies. [78,79] The smoking-induced insulin resistance is also associated with an increase in triglyceride count, [8] because in fatty tissue glucose is converted to triglycerides. In turn, due to increased serum concentrations of FFA and triglycerides, insulin-stimulated glucose transport in skeletal muscle of habitual cigarette smokers is relatively impaired in comparison with non-smokers. [80] Insulin resistance and the increase in triglycerides observed in smokers are significant risk factors for the future development of arteriosclerosis and hence cardiovascular disease. [76,81]
Carbon Monoxide

Carbon monoxide (CO) is produced from the incomplete combustion of carbon-containing substances, such as gasoline and tobacco. [82] The background level of CO in the atmosphere is very low and has little effect on humans, while most of the CO produced by natural or technological processes is oxidized to CO2 in the upper atmosphere. Comparatively, then, the 3-6% CO in cigarette smoke (and the 2-3 times higher concentrations in pipe and cigar smoke) represent considerably higher levels than are normally encountered. [83]
Carbon monoxide exposure has been implicated in the process of atherosclerosis, contributing to the accumulation of cholesterol in the aorta and coronary arteries. [84,85] In addition, CO exposure enhances endothelial damage, leading to detrimental effects in the presence of ischaemic heart or peripheral vascular disease. [86] The deleterious effects of CO are more profound in the myocardium than in peripheral tissues, because of the very high oxygen extraction by the myocardium at rest. [86] There is epidemiological evidence that workers exposed to high CO concentrations have higher cardiovascular morbidity and mortality compared to the expected rate in the general population. [87,88] The main mechanism by which CO causes heart disease is through hypoxia. Inhalation of cigarette smoke, by either active or passive smokers, increases the levels of carboxyhaemoglobin (COHb) in the blood, decreasing the supply of O2 to the tissues. In addition, myoglobin binds with CO so that the heart muscle does not take up the necessary O2 and does not perform optimally. The reduced O2 uptake as a result of smoking, together with an increase in serum lactic acid levels (lactic acidosis), leads to a reduction in peak aerobic capacity and to a significant decrease in maximum O2 uptake (VO2max) (Figure 2).
CO and Haemoglobin
The strong chemical affinity between haemoglobin (Hb) and CO is well-known. It has been estimated that the affinity between Hb and CO is 200 times greater than the affinity between Hb and oxygen (O2). [83] A direct consequence of this difference is the widespread binding of Hb by CO in the blood, the creation of COHb, and a great increase in its serum levels, resulting in a significant decrease in oxygen uptake by peripheral tissues. More specifically, the CO in smoke binds Hb, creating COHb [86] through the following reaction:
HbO2 + CO?COHb + O2
where HbO2 is oxyhaemoglobin. [89]
The presence of COHb in the blood, apart from decreasing its O2 saturation, causes a leftward shift in the O2-Hb dissociation curve, further compounding the state of particulate-induced hypoxaemia. Thus, for the same haemoglobin concentration the O2 supply to the tissues and cells is decreased. [83] In smokers, COHb levels are 5% on average and may reach as much as 10% in heavy smokers. In contrast, in non-smokers COHb levels range between 0.5-2%, depending on their exposure to automobile exhaust. [10,47] More specifically, one and a half hours after smoking, COHb levels range on average between 3.9-4.1%, [90] while elsewhere it has been shown that immediately after smoking COHb levels were around 9%. [82]
The increase in blood COHb levels and the reduced O2 supply to the tissues affect the vascular permeability. [10] The increase in endothelial permeability, together with the injuries to the intima of the arterial wall associated with exposure to CO, leads to sub-endothelial oedema manifested by early atherosclerotic changes, such as fat deposition in the arterial walls. [91] Finally, the presence of CO in the blood is considered responsible for severe anatomical and morphological changes in the myocardium, such as partial or total necrosis of muscle fibrils, and degenerative processes in the mitochondria. [92] These morphological changes are similar to those found in hypoxia. [83,86] Other observations include extra- and intracellular oedema, capillary wall oedema, an increase in the number of ribosomes, and reparative fibrotic changes. [92]
CO and Myoglobin
Myoglobin may combine with CO93 and, like Hb, has a greater affinity (30-50 times) with CO than with O2, intensifying the hypoxaemia of peripheral tissues and especially the active muscles. [89] However, myoglobin binds to one molecule of O2, whereas Hb binds to four. [94] Thus, the negative effects of increased COHb levels are much more striking than those of COMb, effectively reducing both the O2 supply to the tissues and the O2 uptake of working muscles. [95]
CO and lactic acidosis
The term “lactic acidosis” refers to high levels of lactic acid in the blood. The reduced efficiency of the Ο2 transportation and supply system in smokers inhibits mitochondrial function. The exposure of mitochondria to smoking-induced oxidative substances results in damage to the mitochondrial DNA, reducing adenosine triphosphate production in heart and muscle cells. [96] Essentially, smoking disturbs the enzyme activity (adenine nucleotide translocator and mitochondrial superoxide dismutase) in mitochondria that is essential for their proliferation, thus reducing their numbers. Because of this damage, the muscles cannot get the energy they need to function (since they no longer have sufficient mitochondria); they therefore seek energy via another route: anaerobic metabolism. [89,96] The latter process, however, has lactic acid as its final product, so that the quantity of circulating lactic acid increases significantly (lactic acidosis), increasing the blood’s acidity, [54] compromising aerobic tolerance, and impairing exercise capacity.
CO and Exercise Capacity
Smoking even one cigarette can immediately affect physical exercise capacity. [97] The effects of CO, such as the widespread binding of Hb and the reduced arterial O2 blood saturation, the insufficiency of respiratory enzymes, in combination with the binding of myoglobin and the effects of CO on aerobic metabolism, result in dysfunction of the O2 production, transportation, and delivery system, especially during exercise. [83,89] Briefly, the reduced quantities of transported O2 and the decreased O2 supply to and uptake from the active tissues, combined with the binding of myoglobin by CO, significantly decrease maximal oxygen uptake (VO2max) reducing the functional capacity and the performance of the circulatory system (Figure 2).
There is an observable decrease, of around 10%, in the duration of exercise until exhaustion in smokers, which is attributable to a reduction in O2 production in the metabolically active tissues, as a result of arterial O2 desaturation, and to the insufficiency of the O2 transportation, supply and uptake system (Figure 2). [89,97] This impaired exercise tolerance and the decreased maximal exercise capacity have been recorded even in young healthy smokers. [63,98]
Regardless of the underlying mechanism, the effect of smoking on COHb levels is responsible for a leftward shift in the O2 -Hb dissociation curve. [89] Thus, increased COHb levels are able to interfere with the O2 released in the cells in two ways, both of which can reduce VO2max: 1) decreasing the amount of O2 transported in the blood via a reduction in the available receptors on the surface of Hb, and 2) delaying the unloading of O2 into active muscles. The result is a decrease in the effectiveness of myoglobin in unbinding O2 in muscle cells. [95] During exercise, the hypoxaemia due to smoking becomes more apparent at the lactic acidosis threshold, where the deoxygenation of skeletal muscles increases drastically. [89]
Similar effects of smoke on the O2 transportation and supply system are seen in individuals who are not active smokers. [99] Since non-smokers are more vulnerable to CO than smokers, simply being exposed to cigarette smoke may reduce their VO2max. The extent to which VO2max is reduced depends on the amount of CO that smokers inhale. [11,82] Horvath et al. claimed that no significant reduction in VO2maxwas observed until levels of COHb reached or exceeded 4.3%, a level exhibited by most smokers. [100] From the moment COHb levels reach 4.3%, VO2maxdecreases in accordance with the following equation:
VO2max = 0.91(%COHb) + 2.2
Conclusions
Smoking, through its basic ingredients nicotine and CO, increases oxidative stress, endothelial damage and dysfunction, is associated with significantly higher serum concentrations of total cholesterol and triglycerides, reduces the cardioprotective HDL, and by promoting intravascular inflammation represents a significant risk factor for the development of atherosclerosis and cardiovascular disease. In addition, nicotine deregulates cardiac autonomic function, boosts sympathetic activity, and increases HR at rest, while it blunts HR elevation during progressive exercise and lowers the maximum HR that can be achieved. In parallel, the smoking-induced CO binds with haemoglobin and myoglobin, reduces arterial O2 blood saturation, and compromises the efficiency of respiratory enzymes, resulting in dysfunction of the O2 production, transportation and delivery system, especially during exercise; this can substantially reduce the functional capacity and the performance of the circulatory system.
Altogether, smoking is the most important modifiable risk factor for cardiovascular disease, a major risk factor for cardiovascular morbidity and mortality, and is considered to be the leading preventable cause of death in the world.
Acknowledgments
The authors would like to thank Philip Lees, Technical Editor of the Hellenic Journal of Cardiology, for his invaluable editorial assistant with the English text.
Sources of support
Laboratories and equipment were provided by TEI-A
Potential conflicts of interest
None
ANNEX

2732
References
- World Health Organization. Report on the Global Tobacco Epidemic. Geneva, 2008; available at: https://www.who.int/tobacco/mpower/mpowerreport_full_2008.pdf (last day accessed 3 October 2013).
- U.S. Department of Health and Human Services. National Center for Chronic Disease Prevention and Health Promotion. Office on Smoking and Health. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta, 2004; available at: https://www.cdc.gov/tobacco/data_statistics/sgr/2004/complete_report/index.htm (last day accessed 22 September 2013).
- World Health Organization. World Health Report on Reducing Risks and Promoting Healthy Life. Geneva, 2002; available at: https://www.who.int/whr/2002/en/whr02_en.pdf (last day accessed 22 September 2013).
- European Society of Cardiology. Position paper on the “Tobacco Products Directive”. Sophia Antipolis Cedex-France, 2013; available at: https://www.escardio.org/about/Documents/tobacco-products-directive- position-paper.pdf (last day accessed 3 October 2013).
- World Health Organization. Regional Office for Europe. The European Tobacco Control Report 2007. Geneva, 2007; available at: https://www.euro.who.int/data/assets/pdf_file/0005/68117/E89842.pdf (last date accessed 3 October 2013).
- Peto R, Lopez AD, Boreham J, Thun M. Mortality from smoking in developed countries 1950 -2000, 2nd edition: revised June 2006; available at: https://www.ctsu.ox.ac.uk/~tobacco/C0002.pdf (last day accessed 27 March 2013).
- Harvard School of Public Health. The Greek Tobacco Epidemic. Center for Global Tobacco Control. Boston, December 2011; available at: www.smokefreegreece.org (last day accessed 27 March 2013).
- Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J Am Coll Cardiol. 2004; 43:1731-1737.
- U.S. Department of Health and Human Services. Public Health Service. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease. A Report of the Surgeon General. USA, 2010; available at: https://www.surgeongeneral.gov/library/reports/tobaccosmoke/executivesummary.pdf (last day accessed 24 July 2013).
- Bullen CH. Impact of tobacco smoking and smoking cessation on cardiovascular risk and disease. Expert Rev Cardiovasc Ther. 2008; 6(6):883-895.
- USA Institute of Medicine of the National Academies. Secondhand Smoke Exposure and Cardiovascular Effects: Making Sense of the Evidence. Washington DC: The National Academies Press, National Academy of Sciences, 2009; available at: https://www.nap.edu/catalog/12649.html (last day accessed 24 July 2013).
- European Union. Special Eurobarometer on Tobacco. Eurobarometer 332, 2010; available at: https://ec.europa.eu/health/tobacco/docs/ebs332_en.pdf (last day accessed 3 October 2013).
- World Health Organization. World Health Statistics. Geneva, 2011; available at: https://www.who.int/gho/publications/world_health_statistics/2011/en/ (last day accessed 24 September 2013).
- Papathanasiou G, Papandreou M, Galanos A, Kortianou E, Tsepis H, Kalfakakou V, et al. Smoking and physical activity interrelations in health science students. Is smoking associated with physical inactivity in young adults? Hellenic J Cardiol. 2012; 53:17-25.
- Lande RG. Nicotine Addiction. Pathophysiology. Walter Reed Army Medical Center. Department of Psychiatry. Medscape Updated, December 13, 2012; available at: https://emedicine.medscape.com/article/287555-overview#a0104 (last day accessed 27 October 2013).
- Robertson D, Tseng CJ, Appalsamy M. Smoking and mechanisms of cardiovascular control. Am Heart J. 1988; 115:258-262.
- Benowitz NL, Gourlay SG. Cardiovascular Toxicity of Nicotine: Implications for Nicotine Replacement Therapy. J Am Coll Cardiol. 1997; 29:1422–1431.
- Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004; 15:1983–1992.
- Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann NY Acad Sci. 1993; 686:12-27.
- Gusarov I, Shatalin K, Starodubtseva M, Nudler E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science. 2009; 325:1380-1384.
- Barua RS, Ambrose JA, Eales-Reynolds LJ, DeVoe MC, Zervas JG, Saha DC. Dysfunctional endothelial ntric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation. 2001; 104:1905-1910.
- Reinders JH, Brinkman HJ, van Mourik JA, de Groot PG. Cigarette smoke impairs endothelial cell prostacyclin production. Arterioscler Thromb Vasc Biol. 1986; 6:15-23.
- Coceani F. Carbon monoxide in vasoregulation: The promise and the challenge. Circ Res. 2000; 86; 1184-1186.
- Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003; 42:1149–1160.
- Craig WY, Palomaki GE, Haddow JE. Cigarette smoking and serum lipid and lipoprotein concentrations: an analysis of published data. Br Med J. 1989; 298(6676):784-788.
- Ball K, Turner R. Smoking and the heart. The basis for action. Lancet. 1974; 2(7884):822-826.
- McGill HC. The cardiovascular pathology of smoking. Am Heart J. 1988; 115:250-257.
- Gnasso A, Haberbosch W, Schettler G, Schmitz G, Augustin J. Acute influence of smoking on plasma lipoproteins. Klin Wochenschr. 1984; 62(Suppl2):36-42.
- McCall MR, van den Berg JJ, Kuypers FA, Tribble DL, Krauss RM, Knoff LJ, et al. Modification of LCAT activity and HDL structure. New links between cigarette smoke and coronary heart disease risk. Arterioscler Thromb. 1994; 14(2):248-253.
- Gepner AD, Piper ME, Johnson HM, Fiore MC, Baker TB, Stein JH. Effects of smoking and smoking cessation on lipids and lipoproteins: outcomes from a randomized clinical trial. Am Heart J. 2011; 161(1):145-51.
- Mjos OD. Effect of free fatty acids on myocardial function and oxygen consumption in intact dogs. J Clin Invest. 1971; 50:1386-1389.
- Kershbaum A, Bellet S, Dickstein ER, Feinbere LJ. Effect of cigarette smoking and nicotine on serum free fatty acids. Based on a study in the human subject and the experimental animal. Circ Res. 1961; 9:631-638.
- Kershbaum A, Khorsandian R, Caplan RF, Bellet S, Feinberg LJ. The role of catecholamines in the free fatty acid response to cigarette smoking. Circulation. 1963; 28:52.
- M. Zamir Ahmad Akbari, Muhammad Sarwar Bhatti, Muhammad Shakoor. Lipid profile in smoking. JAMC. 2000; 12(3):19-21.
- Friedman GD. Cigarette smoking, cotinine, and blood pressure. Circulation. 1989; 80:1493-1494
- U.S. Department of Health and Human Services. Public Health Service. Office on Smoking and Health. The Health Consequences of Smoking: Cardiovascular Diseases. A Report of the Surgeon General, 1983; available at: https://www.ncbi.nlm.nih.gov/books/NBK44704/ (last day accessed 3 October 2013).
- Levine PH. An acute effect of cigarette smoking on platelet function. A possible link between smoking and arterial thrombosis. Circulation. 1973; 48(3):619-623.
- Ross R, Glomset J, Harker L. Response to injury and atherogenesis. Am J Pathol. 1977; 86(3):675-84.
- Green MS, Jucha E, Luz Y. Blood pressure in smokers and nonsmokers: epidemiologic findings. Am Heart J. 1986; 111(5):932-40.
- Hunter KA, Garlick PJ, Broom I, Anderson SE, McNurlan MA. Effects of smoking and abstention from smoking on fibrinogen synthesis in humans. Clin Sci (Lond). 2001; 100(4):459-465.
- Chu AJ. Tissue Factor, Blood Coagulation, and Beyond: An Overview. Int J Inflam. 2011; 2011:367284. doi: 10.4061/2011/367284. Epub 2011 Sep 20.
- Sambola A, Osende J, Hathcock J, Degen M, Nemerson Y, Fuster V, et al. Role of Risk Factors in the Modulation of Tissue Factor Activity and Blood Thrombogenicity. Circulation. 2003; 107:973-977.
- Asthana A, Johnson HM, Piper ME, Fiore MC, Baker TB, Stein JH. Effects of smoking intensity and cessation on inflammatory markers in a large cohort of active smokers. Am Heart J. 2010; 160(3):458-463.
- Dietrich T, Garcia RI, de Pablo P, Schulze PC, Hoffmann K. The effects of cigarette smoking on C- reactive protein concentrations in men and women and its modification by exogenous oral hormones in women. Eur J Cardiovasc Prev Rehabil. 2007; 14(5):694-700.
- Palatini P. Need for a revision of the normal limits of resting heart rate. Hypertension. 1999; 33:622-625.
- Perret-Guillaume C, Joly L, Benetos A. Heart rate as a risk factor for cardiovascular disease. Prog Cardiovasc Dis. 2009; 52:6-10.
- Benowitz NL. Cigarette smoking and cardiovascular disease pathophysiology and implications for treatment. Prog Cardiovasc Dis. 2003; 46:91-111.
- Lauer MS, Pashkow FJ, Larson MG, Levy D. Association of cigarette smoking with chronotropic incompetence and prognosis in the Framingham Heart Study. Circulation. 1997; 96:897-903.
- Narkiewicz K, van de Borne PJ, Hausberg M, Cooley RL, Winniford MD, Davison DE, et al. Cigarette smoking increases sympathetic outflow in humans. Circulation. 1998; 98:528-534.
- Savonen KP, Lakka TA, Laukkanen JA, Halonen PM, Rauramaa TH, Salonen JT, et al. Heart rate response during exercise test and cardiovascular mortality in middle-aged men. Eur Heart J. 2006; 27:582-588.
- Morshedi-Meibodi A, Larson MG, Levy D, O'Donnell CJ, Vasan RS. Heart rate recovery after treadmill exercise testing and risk of cardiovascular disease events (The Framingham Heart Study). Am J Cardiol. 2002; 90:848-852.
- Myers J, Tan SY, Abella J, Aleti V, Froelicher VF. Comparison of the chronotropic response to exercise and heart rate recovery in predicting cardiovascular mortality. Eur J Cardiovasc Prev Rehabil. 2007; 14:215-221.
- Srivastava R, Blackstone EH, Lauer MS. Association of smoking with abnormal exercise heart rate responses and long-term prognosis in a healthy, population-based cohort. Am J Med. 2000; 109:20-26.
- Guyton AC, Hall JE. Textbook of Medical Physiology. Philadelphia: Elsevier/Saunders; 11th ed, 2006.
- Zhu B, Parmley WW. Hemodynamic and vascular effects of active and passive smoking. Am Heart J. 1995; 130:1270-1275.
- Benowitz NL, Hansson A, Jacob P. Cardiovascular effects of nasal and transdermal nicotine and cigarette. Hypertension. 2002; 39:1107-1112.
- Picciotto MR, Brunzell DH, Caldarone BJ. Effect of nicotine and nicotinic receptors on anxiety and depression. Neuroreport. 2002; 13(9):1097-106.
- Jiloha RC. Biological basis of tobacco addiction: Implications for smoking-cessation treatment. Indian J Psychiatry. 2010; 52(4):301–307.
- Jouven X, Empana JP, Escolano S, Buyck JF, Tafflet M, Desnos M, et al. Relation of heart rate at rest and long-term (>20 Years) death rate in initially healthy middle-aged men. Am J Cardiol. 2009; 103:279-283.
- Cooney MT, Vartiainen E, Laakitainen T, Juolevi A, Dudina A, Graham IM. Elevated resting heart rate is an independent risk factor for cardiovascular disease in healthy men and women. Am Heart J. 2010; 159(4):612-619.e3
- Kobayashi Y, Takeuchi T, Hosoi T, Loeppky JA. Effects of habitual smoking on cardiorespiratory responses to sub-maximal exercise. J Physiol Anthropol Appl Human Sci. 2004; 23:163-169.
- Minami J, Ishimitsu T, Matsuoka H. Effects of smoking cessation on blood pressure and heart rate variability in habitual smokers. Hypertension. 1999; 33:586-590.
- Papathanasiou G, Georgakopoulos D, Papageorgiou E, Zerva E, Michalis L, Kalfakou V, et al. Effects of smoking on heart rate at rest and during exercise, and on heart rate recovery in young adults. Hellenic J Cardiol. 2013; 54:168-177.
- Lucini D, Bertocchi F, Malliani A, Pagani M. A controlled study of the autonomic changes produced by habitual cigarette smoking in healthy subjects. Cardiovasc Res. 1996; 31:633-639.
- Hayano J, Yamada M, Sakakibara Y, Fujinami T, Yokoyama K, Watanabe Y, et al. Short and long-term effects of cigarette smoking on heart rate variability. Am J Cardiol. 1990; 65:84-88.
- Laustiola KE, Lassila R, Kaprio J. Decreased β-adrenergic receptor density and catecholamine response in male cigarette smokers. A study of monozygotic twin pairs discordant for smoking. Circulation. 1988; 78:1234-1240.
- Hering D, Somers VK, Kara T, Kucharska W, Jurak P, Bieniaszewski L, et al. Sympathetic neural responses to smoking are age dependent. J Hypertens. 2006; 24:691-695.
- Astrand PO, Rodahl K, Dahl HA, Stromme SB. Textbook of work physiology. Physiological basis of Exercise. Champagne, IL: Human Kinetics; 2003.
- Lauer MS. Chronotropic incompetence. Ready for prime time. JACC. 2004; 44:431-432.
- Asthana A, Piper ME, McBride PE, Ward A, Fiore MC, Baker TB, et al. Long-term effects of smoking and smoking cessation on exercise stress testing: three-year outcomes from a randomized clinical trial. Am Heart J. 2012; 163(1):81-87.e1.
- Bernaards CM, Twisk JWR, Mechelen WV. A longitudinal study on smoking in relationship to fitness and heart rate response. Med Sci Sports Exerc. 2003; 35:793-800.
- Shetler K, Marcus R, Froelicher VF, Vora S, Kalisetti D, Prakash M, et al. Heart rate recovery: validation and methodologic issues. J Am Coll Cardiol. 2001; 38:1980-1987.
- Morise AP. Heart rate recovery. Predictor of risk today and target of therapy tomorrow? Circulation. 2004; 110:2778-2780.
- Cole CR, Foody JM, Blackstone EH, Lauer MS. Heart rate recovery after submaximal exercise testing as a predictor of mortality in a cardiovascularly healthy cohort. Ann Intern Med. 2000; 132:552-555.
- Vander AJ, Sherman JH, Luciano DS. Human Physiology. The mechanisms of body function. 8th Edition. USA N.Y.: The McGraw-Hill Companies Inc., 2001.
- Chiolero A, Faeh D, Paccaud F, Cornuz J. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. 2008; 87(4):801-809.
- Benowitz NL. Pharmacologic aspects of cigarette smoking and nicotine addiction. N Engl J Med. 1988; 319:1318-1330.
- Assali AR, Beigel Y, Schreibman R, Shafer Z, Fainaru M. Weight gain and insulin resistance during nicotine replacement therapy. Clin Cardiol. 1999; 22(5):357-360.
- Eliasson B, Attvall S, Taskinen MR and Smith U. The insulin resistance syndrome in smokers is related to smoking habits. Arterioscler Thromb Vasc Biol. 1994; 14:1946-1950.
- Rincón J, Krook A, Galuska D, Wallberg-Henriksson H, Zierath JR. Altered skeletal muscle glucose transport and blood lipid levels in habitual cigarette smokers. Clin Physiol. 1999; 19(2):135-142.
- Cena H, Tesone A, Niniano R, Cerveri I, Roggi C, Turconi G. Prevalence rate of Metabolic Syndrome in a group of light and heavy smokers. Diabetol Metab Syndr. 2013; 5(1):28.
- Rietbrock N, Kunkel S, Worner W, Eyer P. Oxygen-dissociation kinetics in the blood of smokers and non-smokers: interaction between oxygen and carbon monoxide and the hemoglobin molecule. Naunyn Schmiedebergs Arch Pharmacol. 1992; 345:123-128.
- Turino GN. Effects of carbon monoxide on the cardiorespiratory system. Circulation 1981; 63:253.
- Astrup P, Kjeldsen K, Wanstrup J. Effects of carbon monoxide exposure on the arterial walls. Ann N Y Acad Sci. 1970; 174(1):294-300.
- Thomsen HD. Carbon monoxide-induced atherosclerosis in primates. An electron-microscopic study on the coronary arteries of Macaca trus monkeys. Atherosclerosis. 1974; 20:233–240.
- Zevin S, Saunders S, Gourlay SG, Jacob P, Benowitz NL. Cardiovascular effects of carbon monoxide and cigarette. J Am Coll Cardiol. 2001; 38:1633-1638.
- Stern FB, Lemen RA, Curtis RA. Exposure of motor vehicle examiners to carbon monoxide: a historical prospective mortality study. Arch Environ Health. 1981; 36:59-65.
- Koskela RS. Cardiovascular diseases among foundry workers exposed to carbon monoxide. Scand J Work Environ Health 1994; 20:286-293.
- McDonough P, Moffatt RJ. Smoking-induced elevations in blood carboxyhaemoglobin levels. Effect on maximal oxygen uptake. Sports Med. 1999; 27:275-283.
- Jarvis MJ, Tunstall-Pedoe H, Feyerabend C, Vesey C, Saloojee Y. Comparison of tests used to distinguish smokers from nonsmokers. Am J Public Health. 1987; 77:1435-1438.
- Kjeldsen K, Astrup P, Wanstrup J. Reversal of rabbit atheromatosis by hyperoxia.
- Astrup P. Carbon monoxide, smoking, and cardiovascular disease. Circulation. 1973; 48;1167-1168.
- Rodwell VW. Proteins: myoglobin and hemoglobin. In: Murray RK, Granner DK, Darryl K, et al. editors. Harper’s Biochemistry. Stamford (CT): Appleton & Lange, 1996: pp.53-63.
- Wittenberg BA, Wittenberg JB. Transport of oxygen in muscle. Ann Rev Physiol. 1989; 51:857-878.
- King CE, Dodd SL, Cain SM. O2 delivery to contracting muscle during hypoxic or CO hypoxia. J Appl Physiol. 1987; 63:726-732.
- Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: Nearly as large as smoking. Circulation. 2005; 111:2684-2698.
- Campaign for Tobacco-Free Kids. United States Edition. Smoking, physical activity and poor physical performance. March 14, 2002. Available at: https://www.tobaccofreekids.org/research/factsheets/pdf/0177.pdf (last day assessed 27 October 2013).
- Papathanasiou G, Georgakopoulos D, Georgoudis G, Spyropoulos P, Perrea D, Evangelou A. Effects of chronic smoking on exercise tolerance and on heart rate systolic blood pressure product in young healthy adults. Eur J Cardiovasc Prev Rehabil. 2007; 14(5):646-652.
- Colberg SR, Casazza GA, Horning MA, Brooks GA. Increased dependence on blood glucose in smokers during rest and sustained exercise. J Appl Physiol. 1994; 76:26-32.
- Horvath SM, Raven PB, Dahms TE, Gray DJ. Maximal aerobic capacity at different levels of carboxyhemoglobin. J Appl Physiol. 1975; 38(2):300-303.