Keywords
Leukemia; Myeloid; Acute; Oncogenes; Protein-tyrosine kinases; Prognosis; Mutations
Metodología de Busqueda
Este estudio tuvo un enfoque de tipo cualitativo, interpretativodescriptivo y un diseño no experimental. Se llevó a cabo una revisión de literatura con documentos bibliográficos especializados publicados en revistas indexadas, en su mayoría. La búsqueda se realizó durante el mes de agosto de 2016, mediante la utilización de las bases de datos PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), Scopus (https://www-scopus-com.itm.elogim.com:2443/home.uri), Web of Knowledge (https://apps.webofknowledge.com.itm.elogim.com/WOS_GeneralSearch_ input.do?product=WOS&search_mode=GeneralSearch&SID=3EP CBQS5JO4r4hpYGjE&preferencesSaved=) y Science Direct (https://www.sciencedirect.com.itm.elogim.com/). Como parámetros de búsqueda se usaron los siguientes términos MESH: FLT3 ligand protein; FLT-3 ligand; fms-like tyrosine kinase 3 ligand; Leukemia, Myeloid, Acute; antineoplastic agents; sunitinib; midostaurin; sorafenib; entre otros. Las búsquedas crudas arrojaban entre 3000-5000 artículos; sin embargo, al cruzar los términos, se obtenían entre 100-200 artículos. Se usaron filtros para depurar las búsquedas, entre ellos que los trabajos fueran hechos en humanos o células humanas y/o que las publicaciones no tuvieran más de 10 años de antigüedad. En algunos casos se incluyeron artículos anteriores a 10 años, cuando la revisión lo ameritaba por el impacto que había tenido dicho artículo. El objetivo de esta revisión, es ofrecerle al profesional de la salud -especialmente estudiantes de medicina y médicos generales- una actualización del papel que tiene esta molécula en el diagnóstico, pronóstico y tratamiento de la leucemia mieloide aguda, y cómo la determinación de este biomarcador puede cambiar el pronóstico de este tipo de pacientes.
Introducción
La leucemia mieloide aguda (LMA), es un trastorno clonal maligno que se caracteriza por alteraciones y baja producción de células hematopoyéticas saludables; estas alteraciones inhiben la diferenciación de las células e inducen a la proliferación o acumulación excesiva de blastos, los cuales reemplazan al tejido hematopoyético normal, provocando citopenias de las tres principales líneas hematológicas [1-3]. Esta enfermedad se asocia con manifestaciones clínicas inespecíficas como fatiga, disnea, anorexia, pérdida de peso e infecciones y hemorragias que suelen estar presentes en el diagnóstico de la LMA y son dominantes durante todo el tratamiento [4,5]. Sin tratamiento, la enfermedad progresa fatalmente debido a sangrado e infecciones [6]. Actualmente, con los tratamientos disponibles, tan sólo el 35- 40% de los pacientes menores de 60 años llegan a la curación de la enfermedad [7].
La LMA representa cerca del 30-35% de los casos de leucemia y es el tipo de leucemia más común en adultos, siendo la edad promedio de diagnóstico de aproximadamente 66 años y la incidencia aumenta progresivamente con la edad [8-10]. Cerca de 190.000 personas son diagnosticadas cada año en todo el mundo, con una incidencia ASR (Age-standardized rate) de 3.5 para hombres y de 2.2 para mujeres. Cerca de 150.000 personas mueren en un año en todo el mundo por LMA [1,10]. Por lo general, la LMA es una enfermedad que afecta a personas de edad avanzada y es muy poco común en personas menores de 45 años.
En Estados Unidos, en donde existen datos completos del comportamiento epidemiológico de esta enfermedad, se presentan aproximadamente 21.000 nuevos casos de LMA anualmente y aproximadamente 10.000 pacientes mueren cada año como consecuencia de la enfermedad [11].
En los últimos treinta años se ha visto una mejoría en el pronóstico de la LMA, siendo atribuido este progreso al refinamiento de tratamientos de soporte, más que al desarrollo de nuevos medicamentos. Sin embargo, más de la mitad de pacientes, entre jóvenes y adultos y aproximadamente el 90% de pacientes mayores de 65 años mueren a causa de la enfermedad. La recaída después de la remisión completa, sigue siendo uno de los principales obstáculos para obtener la curación [4].
Existen algunas lesiones moleculares que se encuentran en pacientes con LMA y cuya presencia es determinante como factor pronóstico, tales como alteraciones genéticas puntuales y anormalidades cromosómicas. Éstas últimas incluyen deleciones, translocaciones e inversiones, que han sido identificadas en aproximadamente el 50% de los pacientes adultos con LMA de novo y son reconocidas como promotoras del desarrollo de esta enfermedad. Los pacientes que tienen alteraciones en los cromosomas 5 y 7, reordenamiento 11q23 o cariotipo con más de tres anormalidades cromosómicas presentan baja respuesta a la terapia y menor sobrevida (entre 3-10% a 5 años), mientras que las alteraciones t(15;17) (q22; q12), inv(16)(p13.1;q22) y t(8;21) (q22;q22) son asociadas con una mayor tasa de remisión y mejor pronóstico [7,12-14].
Por otra parte, la genotipificación de los pacientes con LMA cumple un papel importante en la estratificación de riesgo y la selección de un posible tratamiento [15,16]. Por ejemplo, mutaciones en CEBPA y NPM1, con prevalencias del 6-10% y 25- 35% respectivamente, son factores pronóstico favorables; estas alteraciones bloquean la diferenciación con un efecto mínimo sobre la proliferación [17]. Así mismo, mutaciones en el gen NPM1 son más frecuente en pacientes con citogenética normal y se asocian a un resultado favorable en pacientes jóvenes sin la mutación FLT3-ITD. Además, pacientes mayores de 60 años con NPM1 mutado tienen mejor respuesta a la quimioterapia intensiva convencional. De otra parte, duplicaciones en FLT3, MLL, IDH1/IDH2, KIT, TET2, mutaciones en DNMT3A y sobreexpresión de BAALC, ERG, o MN1 son de mal pronóstico, ya que afectan las vías de señalización proliferativas, lo cual causa el crecimiento anormal de células leucémicas. La incidencia de las mutaciones en los genes IDH2, DNMT3A y TET2 aumentan con la edad; además, las mutaciones en estos dos últimos genes son eventos tempranos de la leucemogénesis [7]. Otras alteraciones moleculares como mutaciones de micro RNAs y metilación alterada de los genes influyen también en del desarrollo de la LMA [18,19].
El Gen FLT3
El proceso de hematopoyesis implica una compleja regulación de la proliferación y diferenciación de diversos tipos de células que componen la médula ósea y la sangre periférica, incluyendo los linajes mieloide y linfoide. El control de la proliferación de las células hematopoyéticas es llevado a cabo, en parte, por la estimulación inducida por ligandos de receptores tirosina quinasa. Uno de estos receptores implicado en este proceso ontogénico hematopoyético es FLT3, y en este sentido, se ha visto que mutaciones en dicho gen, se encuentran presentes en un alto porcentaje de pacientes con LMA y están involucradas como factor pronóstico de los pacientes [20].
La proteína FLT3 pertenece a la familia clase III de receptor tirosina quinasa, la cual incluye miembros estructuralmente similares como c-FMS, c-KIT y el receptor de PDGF, y se expresa principalmente en progenitores mieloides y linfoides, placenta, gónadas y cerebro [21,22]. Tiene un papel importante en la hematopoyesis, a nivel de la diferenciación, proliferación y apoptosis de las células hematopoyéticas [23-25].
FLT3 está localizado en el cromosoma 13q12, y comprende 24 exones (Figura 1), los cuales varían su tamaño desde 83 pb hasta 562 pb, y aproximadamente abarcan 100 kb [26,27].
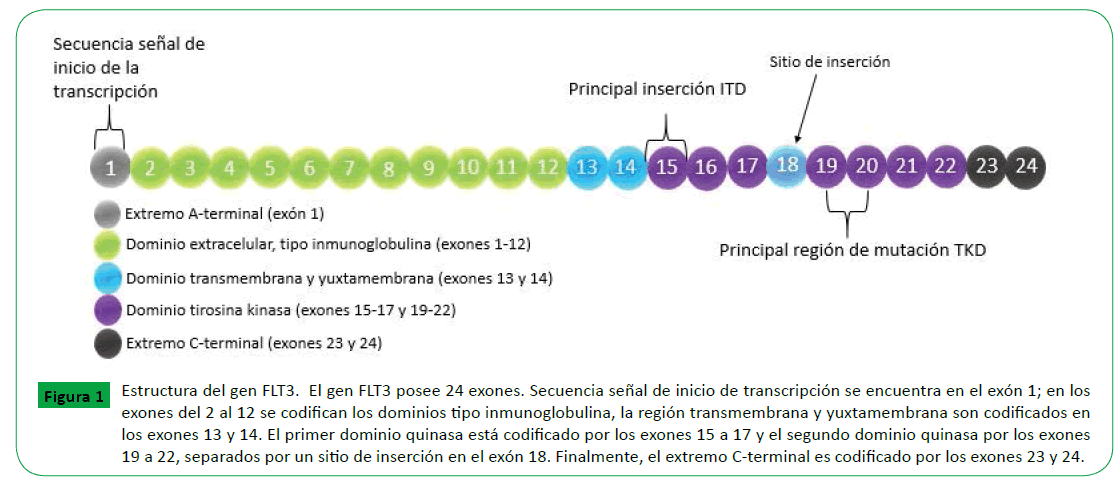
Figura 1: Estructura del gen FLT3. El gen FLT3 posee 24 exones. Secuencia señal de inicio de transcripción se encuentra en el exón 1; en los exones del 2 al 12 se codifican los dominios tipo inmunoglobulina, la región transmembrana y yuxtamembrana son codificados en los exones 13 y 14. El primer dominio quinasa está codificado por los exones 15 a 17 y el segundo dominio quinasa por los exones 19 a 22, separados por un sitio de inserción en el exón 18. Finalmente, el extremo C-terminal es codificado por los exones 23 y 24.
La Proteína FLT3 y su Función
FLT3 codifica una proteína de 993 aminoácidos de longitud [28], la cual posee cinco dominios de tipo inmunoglobulina en la región extracelular, un dominio yuxtamembrana, en donde ocurren las mutaciones ITD (del inglés, Internal Tandem Duplication), dos dominios tirosina quinasa (donde ocurren las mutaciones tipo TKD), separados por un dominio de inserción de quinasa (KI) y un domino C-terminal en la región intracelular [29].
FLT3 se encuentra en forma monomérica no fosforilada. Al interactuar con su ligando, el receptor experimenta un cambio conformacional, formando un homodímero que inicia la activación del dominio tirosina quinasa, conduciendo a la fosforilación de varios sitios en el dominio intracelular [22]. FLT3 activado recluta proteínas en el citoplasma para formar un complejo de interacciones proteína-proteína en el dominio intracelular [30-32].
El gen que codifica el ligando de FLT3, también conocido como FL, fue clonado en 1993 por dos grupos de investigación utilizando una forma soluble del receptor FLT3 [33,34]. Este gen codifica una proteína transmembrana tipo I que contiene un péptido amino-terminal de señalización, cuatro dominios extracelulares helicoidales, espaciador y regiones de amarre, un dominio transmembrana y un pequeño dominio citoplasmático [35]. La actividad del ligando se establece principalmente en los dominios extracelulares, con las regiones espaciadoras y de amarre uniendo el componente extracelular activo al dominio transmembrana, anclando el ligando a la célula [24]. FL es expresado en diferentes tejidos como en el timo, bazo, médula ósea, sangre periférica, ovarios, próstata, placenta, entre otros; presentando altos niveles de expresión en células mononucleares de sangre periférica [36]. Se ha reportado que FL exógeno incrementa la proliferación de blastos no sólo en pacientes que tienen FLT3 de tipo salvaje (WT), sino también en pacientes con las mutaciones FLT3-ITD y FLT3- TKD, lo que sugiere que FL es importante para la estimulación de ambas formas del gen [37].
La activación de FLT3 regula algunos procesos celulares, como el metabolismo de fosfolípidos, la proliferación, la transcripción y la apoptosis, y mediante esta regulación, FLT3 tiene un papel importante en la hematopoyesis normal y el crecimiento celular [38]. La función de FLT3 es dependiente de los factores de crecimiento, tales como Stem Cell Factor e Interleucina 3 [39,40].
FLT3 es expresado en altos niveles en enfermedades como la leucemia mieloide aguda (LMA), en células B precursoras de leucemia linfoblástica aguda (LLA), en una fracción de células T y en leucemia mieloide crónica [41,42]. Se ha encontrado que niveles muy altos de FLT3-WT pueden promover la activación constitutiva del receptor en células malignas [43] y el aumento en la expresión de FLT3 (WT) en blastos puede estar asociada a un peor pronóstico de la enfermedad [44]. Tiene un papel importante pero no absoluto en la hematopoyesis junto con otros factores de crecimiento para estimular la diferenciación y proliferación de células mieloides y linfoides [45].
FLT3 Vía de Señalización
Como se mencionó anteriormente, el receptor FLT3 es activado por la unión del ligando FL al dominio extracelular del receptor, lo que induce la formación de un homodímero de FLT3 en la membrana plasmática y la autofosforilación del mismo en residuos tirosina. Seguidamente, FLT3 activado tiene la capacidad de fosforilar y activar múltiples moléculas que participan en procesos de control de apoptosis, proliferación y diferenciación de células hematopoyéticas en médula ósea [46]. Se ha demostrado que el dominio citoplasmático de FLT3 se asocia físicamente a las proteínas fosfolipasa C gamma 1, fosfoinositol-3-quinasa (PI3K), proteína ligada al factor de crecimiento 2 (Grb2), Shc y la familia de tirosina quinasas Src, propiciando la fosforilación de las mismas. Además, otras proteínas como Vav, CBL, Fyn y Ras también son componentes de la vía de transducción de señal de FLT3 [31,47]. Se ha observado que dichas interacciones pueden modular la activación de las vías de señalización MAPK [48,49], RAS-RAF-MEK-ERK [33,50], y PI3K/Akt [33,51]. Además, existe evidencia de que proporciona resistencia a inhibidores de la vía PI3K/Akt [52].
Las mutaciones en el dominio yuxtamembrana de FLT3 causan la perdida de la función autoinhibitoria, provocando la activación constitutiva de la actividad quinasa de FLT3 y por ende la activación de las vías de señalización proliferativas ya mencionadas [21]. Una de las diferencias que se han descrito, en cuanto a vías de señalización, entre FLT3-WT y FLT3-ITD es que este último activa fuertemente la vía STAT5, importante para el crecimiento celular, lo cual puede explicar el rol de FLT3-ITD en el crecimiento excesivo de células leucémicas [48,53,54]. Adicionalmente, se ha encontrado que la señalización de STAT5 y la activación de RAC1 podrían estar implicadas en el aumento de los niveles de especies reactivas de oxigeno (ROS, por sus siglas en inglés) en células con la mutación FLT3-ITD, provocando inestabilidad genómica y aumento de daño en el ADN, lo que podría ser una importante causa del mal pronóstico que tienen los pacientes con LMA con FLT3-ITD [55].
En ausencia de mutaciones, FLT3 se encuentra fosforilado en más de dos tercios (2/3) de los pacientes con LMA, esto se debe en parte al aumento de los niveles de transcripción de FLT3 que se suelen encontrar en estos pacientes, por lo cual se estaría dando mayor fosforilación de FLT3 y activación de sus vías [44,56].
Mutaciones del Gen FLT3
Mutaciones en el gen FLT3 se han encontrado en pacientes con leucemia linfoblástica aguda (1-3% de los pacientes), mielodisplasia (5-10%) y LMA (15-35%) haciendo de FLT3 uno de los genes frecuentemente mutados [57-59]. Son dos las mutaciones más comunes producidas en FLT3: duplicación en tándem interna (ITD) que se da en el dominio yuxtamembrana y la mutación en el dominio de tirosina quinasa (TKD) [29,60,61].
La mutación ITD, se da en los exones 14 y 15 [62], su longitud va desde 3 hasta 400 pares de bases, aunque puede llegar hasta 1000 pares de bases. Esta mutación se forma a partir de un fragmento duplicado de la secuencia de codificación del dominio yuxtamembrana, que se encuentra entre el dominio transmembrana y el primer dominio de la tirosina quinasa [57,63,64]. La mutación se asocia con mal pronóstico en la supervivencia de pacientes a largo plazo, en términos de sobrevida global y de sobrevida libre de enfermedad [65]. Aproximadamente el 20 al 25% los pacientes con LMA tienen esta mutación, pero es más frecuente en pacientes con cariotipo normal y con la anormalidad cromosómica t(15;17) [66-68].
La mutación TKD se da en el exón 20, provocando la transversión GAT por TAT, lo que genera cambio de un ácido aspártico a tirosina (D835Y) [69]. Aunque es menos común que la mutación ITD, esta mutación promueve la fosforilación constitutiva del receptor y su consecuente activación independiente del ligando [29]. Según estudios realizados, la incidencia de TKD varía entre 5.8% a 7.7% [70]. Aunque no hay resultados significativos, se asocia a una reducción de la sobrevida global de los pacientes y un mayor porcentaje de blastos [29,62,69,71]. Además estas mutaciones podrían representar un mecanismo de resistencia a los inhibidores de FLT3 [28,72,73].
Perspectivas Terapéuticas
Debido a la importancia que tiene FLT3 en el mecanismo de carcinogénesis y su estimulación en la diferenciación y proliferación de células mieloides, se ha vuelto un blanco molecular atractivo para el desarrollo de nuevas terapias y posibles tratamientos [74].
Un número importante de inhibidores de receptores tirosina quinasa han sido estudiados como fármacos para inhibir FLT3 (Tabla 1). Los inhibidores de tirosina kinasa han sido desarrollados para interrumpir la señalización dependiente de este tipo de receptores en un gran número de neoplasias líquidas y sólidas. Estas moléculas compiten por el sitio de unión del ATP dentro del dominio activo de la kinasa, lo que inhibe la habilidad de la proteína para ser fosforilada, y por lo tanto disminuye su actividad. Estudios in vitro han demostrado que estos medicamentos inhiben la fosforilación de FLT3 silvestre y mutado, y también inducen apoptosis in vitro e in vivo.
| |
Nombre |
Código |
Mecanismo de Acción |
Fase de Ensayo Clínico |
Referencia |
| I Generación |
Sunitinib |
SU11248 |
Inhibe KIT, KDR, PDGFR, FLT3 |
Fase I/II trial |
[75] |
| Midostaurina |
PKC412 |
FLT3-ITD and FLT3-TKD kinase |
Fase I/II y III randomizado |
ClinicalTrials.gov identifier: NCT00651261 |
| Lestaurtinib |
CEP701 |
Inhibe JAK2 |
Fase II |
[76] |
| Sorafenib |
|
VEGFR, PDGFR, RAF-1, KIT y FLT3 |
Fase I/II |
[77,78] |
| II Generación |
Gilteritinib |
ASP2215 |
Inhibe FLT3-ITD y FLT3-TKD, también inhibe AXL |
Fase I/II |
[72] |
| Quizartinib |
AC220 |
Inhibe FLT3 mutante, KIT y PDGFRA |
Fase I/II |
[72] |
| Crenolanib |
|
Actividad contra mutaciones en el loop activador de FLT3 (TKD) |
Fase II |
[79] |
Tabla 1 Moleculas inhibidoras de flt3 evaluadas en ensayos clinicos [80].
Inhibidores de FLT3 de Primera Generación
Los inhibidores de FLT3 de primera generación se desarrollaron hace varios años, e incluyen midostaurin, lestaurtinib, sunitinib y sorafenib. Estos medicamentos han sido utilizados como monoterapia o combinados con protocolos estándar de quimioterapia. Estos fármacos son poco específicos para FLT3, siendo efectivos también contra otras quinasas como KIT, PDGFR, VEGFR y JAK2. Estos efectos moleculares colaterales pueden ser responsables tanto de la alta toxicidad, como de la baja eficacia clínica que tienen en pacientes con FLT3 no mutado. Sin embargo, su eficacia también es baja en pacientes con FLT3 mutado que tienen alta carga alélica.
Midostaurin: Tiene actividad contra PKC-alfa, VEGFR, KIT, PDGFR y FLT3. Es activo contra las mutaciones ITD y TKD del gen FLT3. En un estudio fase I, pacientes con LMA y mutación en el gen FLT3, fueron tratados con 75 mg de midostaurin por vía oral 3 veces al día. Se observó que el conteo de blastos periféricos y en médula ósea disminuyó al menos en un 50% de los pacientes [81]. En otro estudio, 95 pacientes con LMA o síndrome mielodisplásico de alto riesgo fueron tratados en un ensayo de fase II con midostaurin oral, con dosis de 50 o 100 mg 2 veces al día. El 71% de los pacientes con mutaciones en FLT3 y el 42% de los pacientes con FLT3 (wt) lograron una reducción de blastos en médula ósea. Las tasas de respuesta y toxicidad no se vieron afectadas por la dosis suministrada [82]. En otro estudio de fase III, 717 pacientes fueron asignados al azar para recibir midostaurina (M) o placebo (P) en la quimioterapia estándar de inducción y de consolidación, seguido por 1 año de mantenimiento con M o P. Los pacientes que fueron tratados con midostaurin mostraron una mejoría de la sobrevida global (HR=0.7) y de la sobrevida libre de evento (HR: 0.8) [83].
Sunitinib: Es un derivado de la indolinona y ha sido probado para tumores estromales gastrointestinales y neuroendocrinos, además de carcinoma de células renales. Inhibe tirosinas quinasas como KIT, PDGFR y tiene mayor especificidad contra FLT3. Parece estar activo contra algunas mutaciones TKD [84,85]. En un estudio fase I, en 15 pacientes con LMA avanzada, se observó una reducción de blastos periféricos en 7 pacientes, sin embargo 2 pacientes murieron por cardiotoxicidad [86]. Recientemente, resultados obtenidos por Fiedler et al [75], lograron demostrar que la adición de sunitinib (25mg/d por 7 días) a la quimioterapia estándar de inducción y consolidación, y su mantenimiento por 2 años, mejora las tasas de remisión completa y los resultados a largo plazo en sobrevida.
Lestaurtinib: Es un inhibidor derivado de indolocarbazol, el cual al igual que la midostaurin, tiene un alto potencial contra las mutaciones FLT3-ITD y TKD [84,87]. Se ha demostrado que lestaurtinib inhibe FLT3-WT y FLT3 mutado en estudios in vitro, y hay un alto grado de correlación entre la inhibición de FLT3 y la citotoxicidad [88]. En un ensayo fase II utilizando lestaurtinib oral como monoterapia durante 8 semanas en pacientes con LMA no tratada, se observó reducciones transitorias de blastos en sangre periférica y en médula ósea en 3 de 5 pacientes con FLT3 mutado y en 5 de 22 pacientes evaluados con FLT3-WT [89].
Sorafenib: Este inhibidor tiene una actividad mucho más específica contra la proteína FLT3-ITD, comparado con la forma silvestre, mientras que tiene baja actividad contra las mutaciones TKD de este gen [87]. La eficacia clínica de este inhibidor se reduce si no se combina con quimioterapia; en un estudio se demostró que hubo una buena tasa de remisión completa en pacientes con LMA refractaria y mutaciones en FLT3 [90]. Sin embargo, en un estudio posterior se reportó que en pacientes ancianos con LMA no se observó este mismo efecto cuando el inhibidor fue combinado con quimioterapia estándar. Es por esto que se requieren más estudios para evaluar la eficacia de la terapia en combinación con este inhibidor [91]. En un ensayo de fase I/II, se combinó citarabina e idarubicina con 400 mg de sorafenib por vía oral dos veces al día en los días 1 a 7 en pacientes menores de 65 años. Diez pacientes fueron tratados en el componente de fase I y 51 pacientes fueron evaluados en fase II. De estos 51 pacientes, el 75% logró remisión completa, incluyendo 14 de 15 pacientes con mutaciones en FLT3 [90,92].
Inhibidores de FLT3 de Segunda Generación
En los estudios de fase temprana de inhibidores de primera generación de FLT3 se encontraron eventos adversos graves, problemas para mantener la concentración plasmática eficaz y baja sensibilidad y efectividad para FLT3. Debido a esto, surgió la necesidad de descubrir y someter a ensayos clínicos nuevos inhibidores que fueran selectivos para FLT3, los que serían clasificados como inhibidores de segunda generación; incluyendo quizartinib, crenolanib y ASP2215, fármacos más potentes y selectivos, con menores IC50 y menos efectos moleculares colaterales.
Quizartinib: Conocido como AC220 y fue desarrollado para tratar mutaciones de FLT3 en LMA, también es inhibidor de otras kinasas como KIT y PDGFRA [93]. Se han realizado una variedad de estudios utilizando quizartinib en LMA en fase de recaída en pacientes jóvenes y ancianos, en los que el aumento de la tasa de remisión completa o parcial osciló entre 61 y 72%. A pesar de la actividad de Quizartinib, el 50% de los pacientes recaen dentro de los 3 meses siguientes [94]. En ensayos fase I/II, la terapia con quizartinib ha sido bien tolerada y ha demostrado un número apreciable de respuestas completas y parciales en pacientes con mutaciones FLT3 [86].
Gilteritinib: También llamado ASP2215, el cual es un potente inhibidor de FLT3 ITD y TKD, tiene además actividad inhibitoria contra Axl. En un estudio fase I/II, con 82 pacientes con LMA en recaída y FLT3 mutado, la tasa de respuesta global en estos pacientes fue del 57%. En 68 pacientes que fueron tratados con dosis de 80 mg o más, la tasa de respuesta global fue de 63% [72].
Crenolanib: Originalmente diseñado para inhibir PDGFR, es también un potente inhibidor de FLT3-ITD, FLT3-TKD y también de las formas silvestres de FLT3. Se ha demostrado eficacia de crenolanib contra líneas celulares tumorales y blastos primarios que han desarrollado mutaciones D835 [95]. En un ensayo fase II que incluyó pacientes con LMA refractaria, crenolanib tuvo una mejor actividad en pacientes no tratados previamente con inhibidores de FLT3 en comparación con pacientes previamente tratados (remisión completa con recuento sanguíneo incompleto 23% vs. 5% respectivamente). El crenolanib también fue probado frente a un panel de líneas celulares mutantes D835 y mostró una citotoxicidad superior cuando se comparó con otros inhibidores de FLT3, tales como quizartinib y sorafenib [96]. En la actualidad se está llevando a cabo un estudio multicéntrico, para evaluar seguridad y tolerancia de crenolanib combinado con quimioterapia de inducción (NCT02283177) [97].
Conclusión
La leucemia mieloide aguda es una enfermedad heterogénea. A pesar de los avances en tratamiento de soporte, la sobrevida a largo plazo sigue siendo baja. El enfoque de la enfermedad desde una perspectiva genética permite entender mejor su fisiopatología, y abordar de una manera más precisa el seguimiento y tratamiento de esta enfermedad. En este sentido, el gen FLT3 es un marcador pronóstico importante en LMA. Las mutaciones en este gen se encuentran frecuentemente en pacientes con LMA y están asociadas a baja supervivencia a largo plazo y menor respuesta al tratamiento. Por esta razón, este gen es considerado un blanco terapéutico interesante y se ha promovido el desarrollo de inhibidores específicos para este gen con el objetivo de mejorar la respuesta a los tratamientos y la expectativa de supervivencia de los pacientes. Es importante el estudio juicioso de estos genes, potenciales marcadores de pronóstico, seguimiento y tratamiento en LMA, ya que una nueva era en el manejo de esta enfermedad está llegando, con novedosos fármacos que permitirán mejores respuestas, con sobrevidas prolongadas, especialmente en pacientes con recaída o enfermedad refractaria, y aquellos con resultados citogenéticos de mal pronóstico.
Declaración de Conflictos
Los autores declaran no tener ningún conflicto de intereses.
Agradecimientos
Agradecemos a Colciencias por la financiación del proyecto 115065743926, convocatoria 657 2014. También al Instituto Tecnológico Metropolitano por el apoyo para la realización del presente manuscrito.
22311
References
- Kantarjian HM, Wolff RA (2016) Adult acute myeloid leukemia. In: Edmonson KG, Pancotti R, (ed.) The MD Anderson Manual of Medical Oncology. (2nd edn.), McGraw-Hill Education.
- Hernández C (2006) Leucemia mieloide aguda: Diagnóstico, estudio y tratamiento. Manual de prácticas médicas - hospital hermanos ameijeiras. La Habana.
- Gomez Raccio A (2009) Mesa redonda: Formas de presentación frecuentes e infrecuentes de las inmunodeficiencias primarias. Casos clínicos. Tema: Anomalías del hemograma. In: 35 Congreso Argentino de Pediatría.
- Ferrara F, Schiffer CA (2013) Acute myeloid leukaemia in adults. Lancet 381: 484-495.
- De Kouchkovsky I, Abdul-Hay M (2016) Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J 6: e441.
- Döhner H, Weisdorf DJ, Bloomfield CD (2015) Acute Myeloid Leukemia. N Engl J Med 373: 1136-1152.
- Deschler B, Lübbert M (2006) Acute myeloid leukemia: Epidemiology and etiology. Cancer 107: 2099-2107.
- Liesveld JL, Lichtman MA (2015) Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, Levi MM, Press OW, et al., (ed.), Williams Hematology, 9e. New York, NY: McGraw-Hill Education.
- Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. (2017) Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015. JAMA Oncol 3: 524.
- Siegel R, Miller K, Jemal A (2015) Cancer statistics , 2015 . CA Cancer J Clin 65: 29.
- Byrd JC, Mro K, Dodge RK, Carroll AJ, Edwards CG, et al. (2002) Pretreatment cytogenetic abnormalities are predictive of induction success , cumulative incidence of relapse , and overall survival in adult patients with de novo acute myeloid leukemia : Results from Cancer and Leukemia Group B ( CALGB 8461 ). Blood 100: 4325-4336.
- Grimwade D, Hills RK, Moorman a V, Walker H, Chatters S, et al. (2010) Refinement of cytogenetic classification in acute myeloid leukaemia: Determination of prognostic significance of rarer recurring chromosomal abnormalities amongst 5,876 younger adult patients treated in the UK Medical Research Council trials. Br J Haematol 3: 17.
- Meyer SC, Levine RL (2014) Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol 15: e382-e394.
- Döhner H, Estey EEH, Amadori S, Appelbaum FRFR, Büchner T, et al. (2010) Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115: 453-474.
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VIV, Paschka P, et al. (2016) Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 374: 2209-2221.
- Deguchi K, Gilliland DG (2002) Cooperativity between mutations in tyrosine kinases and in hematopoietic transcription factors in AML. Leukemia 16: 740-744.
- Filip AA, Libura M, Giebel S, Haus O (2012) Genetic mechanisms and molecular markers of neoplastic transformation in acute myeloid leukemia. In: Witt M, Dawidowska M, Szczepanski T, (ed.) Molecular aspects of hematologic malignancies. Berlin, Heidelberg: Springer Berlin Heidelberg, pp: 29-54.
- Prada-Arismendy J, Arroyave JC, Röthlisberger S (2016) Molecular biomarkers in acute myeloid leukemia. Blood Rev.
- Janke H, Pastore F, Schumacher D, Herold T, Hopfner KP, et al. (2014) Activating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute myeloid leukemia. PLoS One 9: e89560.
- Gary Gilliland D, Griffin JD (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100: 1532-1542.
- Meshinchi S, Appelbaum FR (2009) Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin Cancer Res 15: 4263-4269.
- Yohe S (2015) Molecular genetic markers in acute myeloid leukemia. J Clin Med 4: 460-478.
- Stirewalt DL, Radich JP (2003) The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 3: 650-665.
- Li L, Piloto O, Kim K-T, Ye Z, Nguyen HB, et al. (2007) FLT3/ITD expression increases expansion, survival and entry into cell cycle of human haematopoietic stem/progenitor cells. Br J Haematol 137: 64-75.
- Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, et al. (2001) Genomic structure of human FLT3: Implications for mutational analysis. Br J Haematol 113: 1076-1077.
- Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, et al. (2001) Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: A cancer and leukemia group B study. Cancer Res 61: 7233-7239.
- Alvarado Y, Kantarjian HM, Luthra R, Ravandi F, Borthakur G, et al. (2014) Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer 120: 2142-2149.
- Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, et al. (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97: 2434-2439.
- Dosil M, Wang S, Lemischka IR (1993) Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol Cell Biol 13: 6572-6785.
- Rosnet O, Marchetto S, deLapeyriere O, Birnbaum D (1991) Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene 6: 1641-1650.
- Zhang S, Mantel C, Broxmeyer HE (1999) Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J Leukoc Biol 65: 372-380.
- Hannum C, Culpepper J, Campbell D, McClanahan T, Zurawski S, et al. (1994) Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cell and is encoded by variant RNAs. Nature 368: 643-648.
- Lyman SD, James L, Bos T Vanden, de Vries P, Brasel K, et al. (1993) Molecular cloning of a ligand for the flt3 flk-2 tyrosine kinase receptor: A proliferative factor for primitive hematopoietic cells. Cell 75: 1157-1167.
- Lyman S, Stocking K, Davison B, Fletcher F, Johnson L, et al. (1995) Structural analysis of human and murine flt3 ligand genomic loci. Oncogene 11: 1165-1172.
- Brasel K, Escobar S, Anderberg R, de Vries P, Gruss HJ, et al. (1995) Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia 9: 1212-1218.
- Bruserud Ø, Hovland R, Wergeland L, Huang TS, Gjertsen BT (2003) Flt3-mediated signaling in human acute myelogenous leukemia (AML) blasts: A functional characterization of the effects of Flt3-ligand in AML cell populations with and without genetic Flt3 abnormalities. Haematologica 88: 416-428.
- Ray RJ, Paige CJ, Furlonger C, Lyman SD, Rottapel R (1996) Flt3 ligand supports the differentiation of early B cell progenitors in the presence of interleukin-11 and interleukin-7. Eur J Immunol 26: 1504-1510.
- Rusten BLS, Lyman SD (1996) The FLT3 Ligand Is a Direct and Potent Stimulator of the Growth of Primitive and Committed Human CD34’ Bone Marrow Progenitor Cells In Vitro. Blood 87: 1317-1325.
- Shah A, Smogorzewska E, Hannum C, Crooks G (1996) Flt3 ligand induces proliferation of quiescent human bone marrow CD34+CD38- cells and maintains progenitor cells in vitro. Blood 87: 3563-3570.
- Drexler HG (1996) Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 10: 588-599.
- Rosnet O, Bühring HJ, Marchetto S, Rappold I, Lavagna C, et al. (1996) Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 10: 238-248.
- Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, et al. (2003) Inhibition of FLT3 in MLL. Cancer Cell 3: 173-183.
- Ozeki K, Kiyoi H, Hirose Y, Iwai M, Ninomiya M, et al. (2004) Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood 103: 1901-1908.
- Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP,et al. (1995) Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity 3: 147-161.
- Takahashi S (2011) Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol 4: 13.
- Lavagna-Sévenier C, Marchetto S, Birnbaum D, Rosnet O (1998) FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia 12: 301-310.
- Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, et al. (2000) Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 19: 624-631.
- Takahashi S (2006) Inhibition of the MEK/MAPK signal transduction pathway strongly impairs the growth of Flt3-ITD cells. Am J Hematol 81: 154-155.
- Srinivasa SP, Doshi PD (2002) Extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways cooperate in mediating cytokine-induced proliferation of a leukemic cell line. Leukemia 16: 244253.
- Zhang S, Broxmeyer HE (2000) Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem Biophys Res Commun 277: 195-199.
- Nogami A, Oshikawa G, Okada K, Fukutake S, Umezawa Y, et al. (2015) FLT3-ITD confers resistance to the PI3K/Akt pathway inhibitors by protecting the mTOR/4EBP1/Mcl-1 pathway through STAT5 activation in acute myeloid leukemia. Oncotarget 6: 9189-9205.
- Choudhary C, Schwäble J, Brandts C, Tickenbrock L, Sargin B, et al. (2005) AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 106: 265-273.
- Mizuki M, Schwäble J, Steur C, Choudhary C, Agrawal S, et al. (2003) Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 101: 3164-3173.
- Sallmyr A, Fan J, Datta K, Kim K-T, Grosu D, et al. (2008) Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 111: 3173-3182.
- Zheng R, Small D (2005) Mutant FLT3 signaling contributes to a block in myeloid differentiation. Leuk Lymphoma 46: 1679-1687.
- Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, et al. (2001) The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the United King. Blood 98.
- Abu-Duhier FM, Goodeve AC, Wilson GA, Gari MA, Peake IR, et al. (2000) FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br J Haematol 111: 190-195.
- Lagunas-Rangel FA, Chávez-Valencia V (2017) FLT3–ITD and its current role in acute myeloid leukaemia. Med Oncol 34: 114.
- Ofran Y, Rowe JM (2013) Genetic profiling in acute myeloid leukaemia {box drawings light horizontal} where are we and what is its role in patient management. Br J Haematol 160: 303-320.
- Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, et al. (1996) Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 10: 1911-1918.
- Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. (2002) Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics : A study of the AML Study Group Ulm. Blood 100: 4372-4380.
- Nguyen B, Williams AB, Young DJ, Ma H, Li L, et al. (2017) FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget 8.
- Schnittger S, Schoch C, Dugas M, Kern W, Staib P, et al. (2002) Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100.
- Bienz M, Ludwig M, Mueller BU, Leibundgut EO, Ratschiller D, et al. (2005) Risk Assessment in Patients with Acute Myeloid Leukemia and a Normal Karyotype. Clin Cancer Res 11: 1416-1124.
- Döhner H, Gaidzik VI (2011) Impact of genetic features on treatment decisions in AML. Hematol Educ Progr 2011: 36-42.
- Blau O, Berenstein R, Sindram A, Blau IW (2013) Molecular analysis of different FLT3-ITD mutations in acute myeloid leukemia. Leuk Lymphoma 54: 145-152.
- Gilliland DG (2004) The Molecular basis of leukemia. Hematology 2004: 80-97.
- Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, et al. (2001) Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol 113: 983-988.
- Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S, et al. (2008) Prognostic relevance of FLT3 -TKD mutations in AML : the combination mattersan analysis of 3082 patients Prognostic relevance of FLT3 -TKD mutations in AML : the combination matters — an analysis of 3082 patients. Blood 111: 2527-2537.
- Thiede C, Steudel C, Mohr B, Schaich M, Schaekel U, et al. (2001) Analysis of FLT3-activating mutations in 713 patients with acute myelogenous leukemia (AML): High prevalence in FAB-subtype M5 and identification of subgroups with poor prognosis. Blood 2001: 2994.
- Hassanein M, Almahayni MH, Ahmed SO, Gaballa S, El Fakih R (2016) FLT3 Inhibitors for Treating Acute Myeloid Leukemia. Clin Lymphoma, Myeloma Leuk 16: 543-549.
- Zhang W, Gao C, Konopleva M, Chen Y, Jacamo RO, et al. (2014) Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin Cancer Res 20: 2363-2374.
- Kovalenko M, Gazit A, Bohmer A (1994) Selective platelet-derived growth factor receptor kinase blockers reverse sis-transformation. Cancer Res 62: 185-190.
- Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, et al. (2005) Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 105: 54-60.
- Fischer T, Stone RM, DeAngelo DJ, Galinsky I, Estey E, et al. (2010) Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 28: 4339-4345.
- Stone RM, Mandrekar S, Sanford BL, Geyer S, Bloomfield CD, et al. (2015) The Multi-Kinase Inhibitor Midostaurin (M) Prolongs Survival Compared with Placebo (P) in Combination with Daunorubicin (D)/Cytarabine (C) Induction (ind), High-Dose C Consolidation (consol), and As Maintenance (maint) Therapy in Newly Diagnosed Acute Mye. Blood 126: 6.
- Kiyoi H (2015) Flt3 Inhibitors: Recent advances and problems for clinical application. Nagoya J Med Sci 77: 7-17.
- Kancha RK, Grundler R, Peschel C, Duyster J (2007) Sensitivity toward sorafenib and sunitinib varies between different activating and drug-resistant FLT3-ITD mutations. Exp Hematol 35: 1522-1526.
- Fiedler W, Serve H, Döhner H, Schwittay M, Ottmann OG, et al. (2005) A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia ( AML ) or not amenable to conventional therapy for the disease. Blood 105: 986-993.
- Fiedler W, Kayser S, Kebenko M, Janning M, Krauter J, et al. (2015) A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol 169: 694-700.
- Grunwald MR, Levis MJ (2013) FLT3 inhibitors for acute myeloid leukemia: A review of their efficacy and mechanisms of resistance. Int J Hematol 97: 683-694.
- Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, et al. (2002) A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 99: 3885-3891.
- Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, et al. (2006) A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 108: 3262-3270.
- Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, et al. (2010) Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol Am Soc Clin Oncol 28: 1856-1862.
- Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, et al. (2013) Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: Results from a randomized, placebo-controlled trial. J Clin Oncol 31: 3110-3118.
- Macdonald DA, Assouline SE, Brandwein J, Kamel-Reid S, Eisenhauer EA, et al. (2013) A phase I/II study of sorafenib in combination with low dose cytarabine in elderly patients with acute myeloid leukemia or high-risk myelodysplastic syndrome from the National Cancer Institute of Canada Clinical Trials Group: trial IND.186. Leuk Lymphoma 54: 760-766.
- Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, et al. (2009) AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 114: 2984-2992.
- Levis MJ, Perl AE, Dombret H, Döhner H, Steffen B, et al. (2012) Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (ac220) in patients with flt3-itd positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. Blood 120: 673.
- Cortes JE, Perl AE, Dombret H, Kayser S, Steffen B, et al. (2012) Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥ 60 years of age with flt3 itd positive or negative relapsed/refractory acute myeloid leukemia. Blood 120: 48.
- Fathi AT (2013) Emergence of crenolanib for FLT3-mutant AML. Blood 122: 3547-3548.
- Randhawa JK, Kantarjian HM, Borthakur G, Thompson PA, Konopleva M, et al. (2014) Results of a phase ii study of crenolanib in relapsed/refractory acute myeloid leukemia patients (pts) with activating flt3 mutations. Blood 124: 389.
- Arog Pharmaceuticals (2014) A safety and tolerability study of crenolanib in combination with chemotherapy in newly diagnosed acute myeloid leukemia patients with flt3 mutations.
- Levis M, Ravandi F, Wang ES, Baer MR, Perl A, et al. (2011) Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 117: 3294-3301.
- Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, et al. (2013) Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 121: 4655-4662.
- Fathi AT, Chen YB (2017) The role of FLT3 inhibitors in the treatment of FLT3-mutated acute myeloid leukemia. Eur J Haematol 98: 330-336.
- Wander SA, Levis MJ, Fathi AT (2014) The evolving role of FLT3 inhibitors in acute myeloid leukemia: Quizartinib and beyond. Ther Adv Hematol 5: 65-77.

