Key words
Creutzfeldt-Jakob’s disease, prion encephalopathy, prion
Caso Clínico
Antecedentes
Hombre de 58 años, trabaja como encargado de obras, cursó estudios básicos, diestro. Vive sólo, separado. No factores de riesgo vascular; bebedor importante hasta el comienzo de la enfermedad actual. No tratamientos crónicos. No antecedentes familiares de enfermedades neurológicas.
Enfermedad actual
Paciente que debuta con síndrome general con astenia, artralgias y pérdida de peso (~10kg. en 3 meses). Además el paciente se encuentra con estado de ánimo deprimido que inicialmente se puso en relación con síndrome reactivo a un problema familiar. Sin embargo, en los meses siguientes hay una evidente progresión de la clínica con desorientación marcada, pérdida de memoria, alteración del sueño, astenia, agitación, desinhibición y cefalea. El paciente es estudiado por Medicina Interna sin hallazgos de interés y finalmente ingresa en una clínica psiquiátrica donde deja de beber alcohol y se instaura tratamiento con antidepresivos, neurolépticos y terapia electroconvulsiva a pesar de lo cual se produce un agravamiento notable: no reconoce a familiares y muestra dificultad fluctuante para expresarse, delirios, fabulaciones y alucinaciones e ilusiones visuales y auditivas.
Exploración
Ingresa entonces en Neurología con la siguiente exploración: buen nivel de conciencia, ansioso, inatento, pupilas, motórica ocular, campimetría, pares craneales, vías largas, pruebas cerebelosas, reflejos y marcha: sin alteraciones; no meníngeos, afebril; exploración general: normal. En la evaluación neuropsicológica (Figura 1) el paciente está desorientado en tiempo, espacio y persona, no recuerda datos como su número de teléfono, dirección…, muestra dificultad para la comprensión verbal oral y escrita así como para expresarse con discurso incoherente y lenguaje espontáneo escaso; repetición preservada; desorganización de ideas; dificultad para reconocer caras. En los test practicados alcanzaba las siguientes puntuaciones: MMSE: 13/30 (Falla en orientación temporal, espacial, cálculo, recuerdo de las palabras, escritura y dibujo); Test 7 Minutos: 3/99 (Test de orientación: 0, Test de memoria: 3, Fluidez categorial: 3, Test del reloj: 0.); Subtest del Barcelona: test de praxias manuales: en general conserva buena destreza manipulativa, cierta dificultad en secuencias bimanuales; test de gnosias (auditivas, colores, estereognosia): no déficit; test de memoria: afectación severa de la de memoria inmediata y de evocación.

Figura 1. Detalles de la exploración neuropsicológica: A. El paciente copia la figura (aunque en lugar de dos pentágonos dibuja un pentágono y un cuadrado); cuando se le pide que escriba su nombre puede hacerlo pero persevera con la letra inicial "J". B. Cuando se le pide que copie la figura lo hace mal, terminando el ejercicio prematuramente porque según refiere "está cansado..." y fabula diciendo que "estuvo estos días cargando maderos". C y D. Es incapaz de dibujar un reloj y colocar las manecillas en la hora propuesta.
Pruebas complementarias
Análisis de sangre: Hemograma, VSG, bioquímica general: normal. Hormonas tiroideas: normales. Anticuerpos antinucleares y transglutaminasa: negativos. HLA: DQA10501 +, DQB10201 +.
Análisis de líquido cefalorraquídeo: proteínas 51, 3 mononucleares, glucosa normal. Proteína priónica positiva. Serologías: IgG positiva con IgM negativa para CMV, varicela, virus ECHO, virus Coxackie. Negativas para virus de Epstein-Barr, Lyme, Brucela, Lues y Herpes.
Gastroscopia y biopsia de mucosa gástrica: gastritis antral Neuroimagen (Figura 2): TC cráneo: atrofia córticosubcortical. Hiperdensidad en cuerpo calloso. RM cráneo: imagen redondeada en el cuerpo calloso sugestiva de angioma venoso. Hiperseñal cortical difusa parcheada de predominio en regiones posteriores apreciable en difusión.
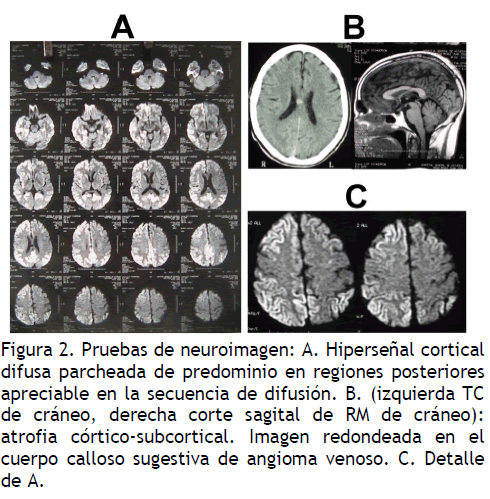
Figura 2. Pruebas de neuroimagen: A. Hiperseñal cortical difusa parcheada de predominio en regiones posteriores apreciable en la secuencia de difusión. B. (izquierda TC de cráneo, derecha corte sagital de RM de cráneo): atrofia córtico-subcortical. Imagen redondeada en el cuerpo calloso sugestiva de angioma venoso. C. Detalle de A.
SPECT cerebral: ligera hipoperfusión frontal inferior de predominio derecho y parietal bilateral. EEGs seriados: inicialmente ritmo de base irregular a 5 Hz; en sucesivos actividad basal más lenta y desorganizada, intercalándose brotes de ondas agudas sin ritmo claro (Figura 3).
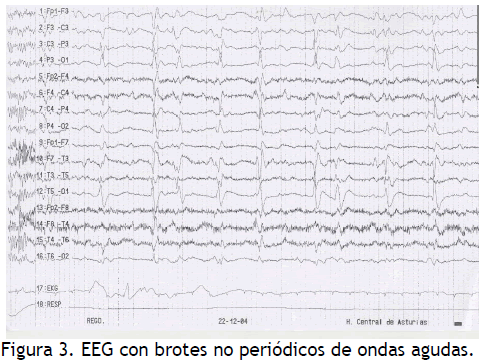
Figura 3. EEG con brotes no periódicos de ondas agudas.
Estudio del gen PRNP: La secuencia del exón 3 coincide con la normal (GENBANK NM000311). El paciente es homocigoto Met para el polimorfismo de la posición 129 (Val/Met).
Evolución
Durante el ingreso comenzó a quejarse de cefalea y episodios de dolor abdominal con diarreas ocasionales. Se mostraba muy inquieto con tendencia a la huída. En la exploración los reflejos osteotendinosos se hicieron más vivos, sin apreciarse mioclonías aunque sí alguna fasciculación difusa. Al alta reducción progresiva del lenguaje con tendencia al “aislamiento”, trastorno del comportamiento y alimentación. Importantes cambios de humor. Rigidez y bradicinesia en las 4 extremidades. Actitud corporal en flexión. Mioclonías reflejas. Incontinencia de esfínteres y en las últimas semanas nivel de conciencia fluctuante llegando al coma y finalmente al exitus a los 20 meses del inicio del cuadro.
Estudio anatomopatológico
Macroscópico: externo normal; al corte discreta atrofia cortical en lóbulos frontales, primera y segunda circunvoluciones temporales, vermix y hemisferios cerebelosos (Figura 4).
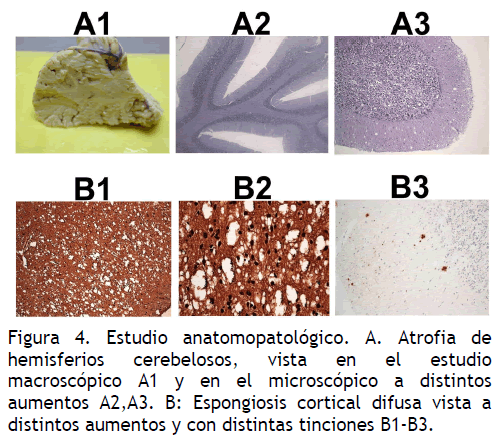
Figura 4. Estudio anatomopatológico. A. Atrofia de hemisferios cerebelosos, vista en el estudio macroscópico A1 y en el microscópico a distintos aumentos A2,A3. B: Espongiosis cortical difusa vista a distintos aumentos y con distintas tinciones B1-B3.
Microscópico: espongiosis difusa, más marcada en corteza frontal, temporal y occipital. Gliosis reactiva en corteza cerebral y cerebelosa. Degeneración vacuolar en ganglios basales y tálamo (Figura 4).
Discusión
Las encefalopatías priónicas se caracterizan por el depósito en el cerebro de una forma anormal, resistente a las proteasas, de una glucoproteína de membrana denominada proteína priónica. Desde el punto de vista anatomopatológico se caracterizan por la presencia de pérdida neuronal, hiperplasia, proliferación de astrocitos, vacuolización del neuropilo (cambios espongiformes) y depósitos de proteína priónica patológica sin reacción inflamatoria. La Enfermedad de Creutzfeldt-Jakob [ECJ] fue descrita en 1920 por los autores que le dan nombre como una demencia rápidamente progresiva acompañada de mioclonías, ataxia y mutismo. A pesar de ser la más común de las encefalopatías espongiformes su incidencia es baja y aún hay muchos aspectos de la misma que resultan poco conocidos. A propósito de este caso trataremos de subrayar alguno de estos aspectos. Respecto a la clínica de inicio decir que puede ser muy variada. En este caso se presentó de forma larvada e inespecífica con un síndrome general acompañado de insomnio y síndrome depresivo. Este tipo de presentación no es infrecuente encontrándose en al menos una cuarta parte de los pacientes [1], por lo que no es raro que estos pacientes sean estudiados por otros especialistas (Internista, Psiquiatría...) antes de llegar al neurólogo. Sin embargo la forma más común es el síndrome que combina ya desde el inicio demencia progresiva con mioclonus, acompañado o no de signos cerebelosos y síntomas psiquiátricos [2]. Formas más raras son las que al debut presentan hipoacusia [3] o síntomas visuales (Variante de Heidenhain con ceguera cortical y alucinaciones visuales) [4,5].
Resulta interesante que el polimorfismo Val/Met en el codón 129 del gen PRNP influencia la expresión clínica de la enfermedad en diferentes aspectos: a) la susceptibilidad a la infección, siendo la homocigosis, especialmente para metionina un factor de riesgo, b) se asocia con curso clínico más rápido, c) mayor edad de presentación y d) ciertos patrones clínicos [1].
También resulta curioso que las distintas cepas pueden transmitir la enfermedad con variaciones en el tiempo de incubación, topografía de la lesión, tipo de lesión y patrón de depósito priónico. Las distintas cepas vienen determinadas por las propiedades fisicoquímicas del prión, como el grado de glicosilación y el tamaño del producto de la proteolisis parcial, que permiten identificar las diferentes cepas. La combinación, por tanto, del genotipo PRNP y la cepa regulará la incidencia de la enfermedad, el tiempo de incubación y el fenotipo [6].
La exploración neuropsicológica suele ser florida pero muy variable y poco específica. Se ha visto que en fases previas a la demencia en algunos casos la exploración neuropsicológica puede evidenciar una afectación órbito-mesial, similar a la encontrada en los pacientes con parálisis supranuclear progresiva [7].
La evolución de la enfermedad es rápidamente progresiva; estimándose un periodo de enfermedad desde el inicio de la sintomatología hasta el fallecimiento del paciente, de entre 2 y 12 meses [8]. Sin embargo existen casos, como el que aquí se presenta, en los que la evolución es algo más lenta, pudiendo superar los 20 meses.
Recientemente se han descrito en pacientes con ECJ alteraciones en la RM cerebral en las secuencias ponderadas en difusión, sobre todo aumento de la señal en los ganglios basales y cortical [9,10]. Son pocos los casos descritos en la bibliografía en los que exista hiperintensidad cortical difusa y parcheada sin alteración de señal en los ganglios basales [11] como ocurrió en nuestro paciente. Esta cuestión es importante, pues es probable que la mayor ventaja de las secuencias de difusión frente a las secuencias T2 o FLAIR sea su mayor sensibilidad para la detección de alteraciones corticales. Las secuencias de difusión se basan en la modulación de la intensidad de señal causada por la difusión de las moléculas de agua. Los valores del coeficiente de difusión son la expresión de la alteración en la difusión del agua en el parénquima cerebral. Parece ser que la alteración de la señal en difusión observada en los pacientes con ECJ se puede corresponder histopatológicamente con la pérdida de la citoarquitectura, la pérdida neuronal y los cambios espongiformes [11]. La difusión de las moléculas de agua podría estar reducida por la compartimentación en vacuolas que se observan en el denominado cambio espongiforme. Se han realizado estudios para comparar en un mismo paciente los hallazgos neuropatológicos con las alteraciones de señal corticales presentes en la RM; algunos no han encotrado correlación [12] mientras que otros sí [11].
1282
References
- Moreno MJ, Romero J. La enfermedad de Creutzfeldt-Jakob esporádica: variabilidad fenotípica. Neurología 2002;17(7):366-77
- Silva AM, Pires MM, Leite AJ, Honavar M, Mendes A, Correia M, Nora M, Silva MR, Costa M, Guimaraes A, Monteiro L. A retrospective study of Creutzfeldt-Jakob disease in North of Portugal 1993-2002: demographic, clinical and neuropathological features. Arq Neuropsiquiatr. 2003;61(4):950-6
- Krishna P, Bauer C. Hearing loss as the initial presentation of Creutzfeldt-Jakob disease. Ear Nose Throat J. 2004;83(8):535-540
- Staffen W, Trinka E, Iglseder B, Pilz P, Homann N, Ladurner G. Clinical and diagnostic findings in a patient with Creutzfeldt-Jakob disease (type Heidenhain). J Neuroimaging. 1997;7(1):50-4.
- Cooper SA, Murray KL, Heath CA, Will RG, Knight RS.Isolated visual symptoms at onset in sporadic Creutzfeldt-Jakob disease: the clinical phenotype of the "Heidenhain variant". Br J Ophthalmol. 2005;89(10):1341-2
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol;39(6):767-78.
- Zarei M, Nouraei SA, Caine D, Hodges JR, Carpenter RH.Neuropsychological and quantitative oculometric study of a case of sporadic Creutzfeldt-Jakob disease at predementia stage. J Neurol Neurosurg Psychiatry. 2002;73(1):56-8
- Menendez-Gonzalez M, Garcia-Fernandez C, Suarez-San Martin E, Anton-Gonzalez C, Blazquez- Menes B. Cronopatología de las enfermedades priónicas. Rev Neurol. 2004;39(10):962-5.
- Rodriguez-Uranga JJ, Gil-Neciga E, Pinero P, Serrano-Pozo A.Diffusion-weighted magnetic resonance sequences in the early diagnosis of Creutzfeldt-Jakob disease. Rev Neurol. 2005 Sep 16-30;41(6):378.
- Tomita I, Sato K, Shirabe S, Nagasato K, Satoh A, Tsujihata M. Serial diffusion-weighted MRI (DWI) in a patient with sporadic Creuztfeldt-Jakob disease. Rinsho Shinkeigaku. 2004;44(3):182-6
- Moreno-Izco F, Martinez-Gil A. Creutzfeldt- Jakob disease: alterations in an isolated cortical signal in diffusion magnetic resonance imaging. Rev Neurol. 2005;40(1):38-42
- Russmann H, Vingerhoets F, Miklossy J, Maeder P, Glatzel M, Aguzzi A, Bogousslavsky J. Sporadic Creutzfeldt-Jakob disease A comparison of pathological findings and diffusion weighted imaging. J Neurol. 2005;252(3):338-42

