Keywords
Estrogens; Endometriosis; Endometrial neoplasia; Carcinogenesis; Endometrial cancer; Obesogenic environment
Introduction
Alteration of hormonal action is not a naturally occurring new phenomenon; for instance, many plants make compounds (phytoestrogens) that mimic estrogenic sex steroids in vertebrates. What is new and yet intractable is the hypothesis of the 'obesogenic environment' that these chemicals create; thus, they have been termed xenoestrogens or 'estrogens' or 'obesogens' in a sense that obesity precedes endometrial cancer. The search for hormones and phytoestrogens drove mid-20th century research that first isolated, and then reproduced, the natural estrogens, progestins, and androgens. Commercial prospects for supplements or contraceptives fueled production of the diethylstilbestrol (DES) in 1938, ethinyl estradiol in 1950s and other synthetic estrogens.
Evidence for 'estrogens' endocrine disruption
Many environmental chemicals have been shown to elicit toxicity by binding to the estrogen receptor and stimulating transcription of estrogen-responsive genes. The estrogenicity of many environmental contaminants may be eliciting adverse effects on wildlife and human populations. For instance, remarks reported by observation of endocrine-disrupting effects of some Polychlorinated Biphenyl (PCB) isomers in vertebrates include altered sex ratios in turtles [1], reduced male rat fertility following early postnatal exposure [2] and altered expression of steroid hormone-metabolizing enzymes [3]. A most vivid example of toxicity associated with alterations in endocrine function is the alligator population level in Lake Apopka, Florida, which has declined steadily over the past decade, despite the maintenance of stable populations in other Florida lakes [4]. Female alligators sampled from the lake exhibited abnormal ovarian morphology [5]. On human hormonal discrepancies, estrogenic or antiestrogenic effects of environmental contaminants have been implicated, too. Breast cancer incidence has increased steadily since the 1940s [6]. Risk of breast cancer increases with increased cumulative estrogen exposure [7]. An epidemiological study linked breast cancer incidence with serum levels of the DDT metabolite DDE [8]. Such observations led to the hypothesis that environmental estrogens are a preventable cause of breast cancer [9]. Endometriosis, the extrauterine growth and proliferation of endometrial cells, is estimated to affect over 6 million women of reproductive age in the USA alone [10]. The etiology of the disease is unknown, but likely involves hormonal influences. A study of rhesus monkeys chronically treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) demonstrated a dose-dependent increase in the incidence of this disease [11]. TCDD can elicit a variety of endocrine-disrupting effects, including reduced estrogen responsiveness [12]. This compound and related halogenated aromatic hydrocarbons are ubiquitous environmental contaminants that may contribute to the incidence of endometriosis among humans. Residents of Seveso, Italy, who were exposed to high levels of TCDD following a chemical plant explosion in 1976 have been identified as viable candidates to assess the relationship between TCDD exposure and endometriosis in humans [13] and may provide insights into the environmental etiology of this condition.
Mechanism of carcinogenesis: 'Estrogens' and the hormonal hypothesis
One of the most intractable biological problems is finding an accurate definition of cancer. Virchow is often quoted as having said 'no man, even under torture, can say exactly what a tumor is' as reported by Uwing in 1916 [14]. Biologically speaking, 'neoplasia' is a better term than cancer or tumor because it denotes an accumulation of cells different from that of 'hyperplasia'. Much later on, by Willis in 1967 [15] proposed a definition in the context of behavior of a neoplasm or by that of a normal cell type− 'a tumor is an abnormal mass of tissue, the growth of which exceeds and is uncoordinated with that of the normal tissues, and persists in the same excessive manner after cessation of the stimuli which evoked the change'. A drawback of Willis’s definition is that it uses the word growth as a synonym of 'cell proliferation'; A second one is that it does not add up the phenomenon of regression that occurs in hormonal carcinogenesis after withdrawal of the hormonal influence.
It is difficult to establish the role played by hormones in the development of neoplasias. It is postulated that hormones play a dualistic role; firstly, that they induce mutations and secondly that they act as promoters; if xenoestrogens act by altering the metabolism of endogenous estrogens, their mutagenic activity would not be associated from the estrogenic activity and linked, instead, with the ability of these xenoestrogens to induce or activate enzymes that regulate the metabolism of endogenous estrogens. A prediction of the mutagenic hypothesis is that only those estrogenic compounds that are mutagenic are expected to induce tumor formation. For instance, 2−fluoroestradiol, a compound with an estrogenic potency similar to estradiol, does not induce tumorigenesis in the Syrian hamster model, while estradiol does; this could be explained by the ability of estradiol to be metabolized to 2−hydroxy metabolites, while 2−fluoroestradiol is not metabolized [16]. From a similar point of view, DES is metabolized to an unstable semiquinone that can react with DNA [17].
Once the tumor develops, it may or may not require hormones to propagate further (hormone sensitive or insensitive); identical to this view, a cell mutated as to its proliferation ability would acquire a selective advantage to proliferate over those untouched to the carcinogen mutagen. A possible explanation could be that hormones play a role in the propagation of hormone-sensitive tumors. It is no coincidence that breast and prostate cancers in human’s regress after estrogens or androgens, respectively. In animal models, regression may cure the tumor. In humans, clinical regression is temporary possibly due to epigenetic mechanisms i.e. short regimens [18,19].
Impact of 'estrogens' on endometrial cancer
Endometrial cancer is the sixth most common cancer in women worldwide and the most common gynecologic malignancy in the developed world [20]. It is estimated that approximately $2.3 billion is spent in the USA each year on endometrial cancer treatment [21]. Most epidemiologic studies have analyzed all types of endometrial carcinoma as a single entity, rather than consider different histopathologic types separately. Nevertheless, historical observations have suggested that endometrial carcinomas vary in histopathologic appearance and clinical features. Based on clinicopathologic observations in 366 endometrial cancers, Bokhman in 1983 proposed that there are two main types of endometrial carcinomas: type 1 tumors related to hormonal imbalances and type 2 tumors that seem largely unrelated to estrogen. According to this model, type 1 tumors are indolent neoplasms that are associated with hyperlipidemia, obesity, and signs of hyperestrogenism, such as anovulatory bleeding, infertility, late menopause, and endometrial and ovarian stromal hyperplasia [22]. Type 2 tumors are unrelated to these features, behave aggressively, and lack the progesterone responsiveness of type 1 tumors. Building on these clinical observations, it has been suggested that the majority of type 1 tumors behave like the endometrioid type of endometrial carcinoma, whereas type 2 tumors probably refer to most serous carcinomas and some other aggressive types [23].
The predominant hypothesis is as follows: unopposed estrogen is the major risk factor for endometrial cancer (Figure 1); this conclusion is based on several observations, including the enhanced rate of endometrial cell proliferation during the follicular phase of the menstrual cycle when estrogen levels are high (and progesterone levels are low) and that women who have used estrogen replacement therapy are much more likely to develop endometrial cancer than women who have never used estrogen replacement therapy [24].
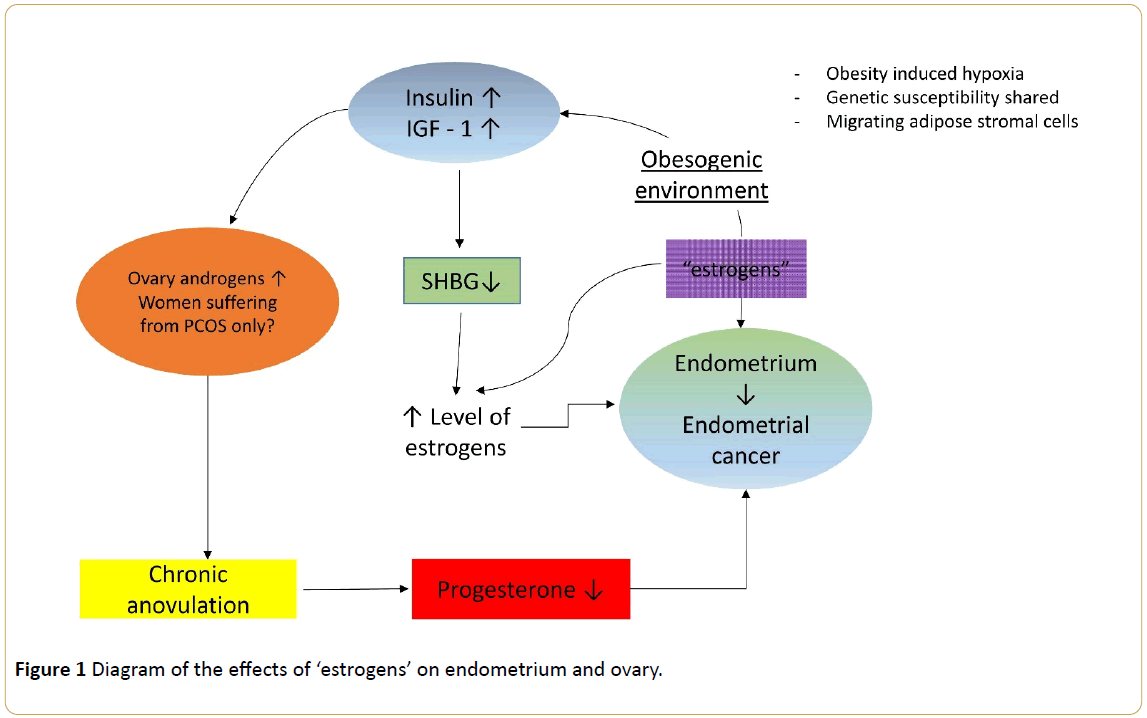
Figure 1: Diagram of the effects of ‘estrogens’ on endometrium and ovary.
Discussion
Menstrual factors, such as early menarche and late menopause, and nulliparity are thought to increase cumulative estrogen exposure by increasing a woman's total lifetime number of menstrual cycles. In polycystic ovary disease, which is characterized by virilization, it is postulated that chronically elevated luteinizing hormone levels promote increased androstenedione production by the ovary, which in turn is converted to estrone, a weker form of estrogen, in peripheral tissue stores [25]. Most endometrioid carcinomas are associated with endometrial hyperplasia. In contrast, almost all serous carcinomas develop from endometrial intraepithelial carcinoma in a background of atrophy [26].
Outbred female mice were treated on days 1-5 with subcutaneous injections of bisphenol A (BPA) (10, 100 or 1000 microg/kg/day) dissolved in corn oil or corn oil alone [27]. At 18 months, ovaries and reproductive tract tissues were examined. There was a statistically significant increase in cystic ovaries and cystic endometrial hyperplasia; severe pathologies of the uterus following neonatal BPA treatment included adenomyosis, leiomyomas, atypical hyperplasia, and stromal polyps [27]. The role of estrogen and the estrogen receptor (ER) in the induction and promotion of tumors was investigated by using transgenic MT-mER mice, which overexpress the ER. It was hypothesized that because of this abnormal expression of the ER, the reproductive-tract tissues of the MT-mER mice may be more susceptible to tumors after neonatal exposure to the potent synthetic estrogen DES [28]. The tumors of the MT-mER mice were often more aggressive than those in the wild-type animals; preceded at 4 months by a significantly higher incidence of the preneoplastic lesion atypical hyperplasia in the MT-mER mice [28], highly indicating that the level of ER present in a tissue may also be a determining factor in the development of estrogen-responsive tumors.
In fact, the lack of DES-effects on gene expression and tissue differentiation, in the estrogen receptor alpha knockout mice uterus, provides evidence of an absolutely necessary role for ER alpha in mediating the detrimental actions of neonatal DES exposure in the murine reproductive tract [29]. Also, it was indicated that neonatal DES exposure temporarily alters expression of multiple chromatin-modifying proteins and persistently alters epigenetic marks in the adult mouse uterus at the Six-1 locus, suggesting a mechanism for developmental exposures leading to altered reproductive function and increased cancer risk [30]. As to other examples of estrogenicity exerted by 'estrogens', triclosan increased the ethinyl estradiol-induced stimulation of epithelial cell height following cotreatment. Cotreatment also enhanced the estrogen-induced change in gene expression, which was reversed with an ER antagonist [31]. The modes of action had been accepted as major pathways for uterine carcinogenesis in rodents are estrogenic activity, increased serum 17betaestradiol (E2) to progesterone ratio and modulation of estrogen metabolism to produce 4-hydroxyestradiol via P450 induction. Concerning uterine adenocarcinoma in rats, cyenopyrafen and benthiavalicarb-isopropyl were predicted to be modulation of estrogen metabolism, while those of pyriminobac-methyl and spirodiclofen were predicted to be increased E2 to Progesterone ratio [32].
The estrogenicity of environmental compounds known as 'estrogens' and its particular role in the development of uterine neoplasia was described in humans. Uterine fibroids are the most frequent gynecologic tumor, affecting 70% to 80% of women over their lifetime [33]. Jeong et al., in order to investigate the number of leiomyoma patients-exposed to BPA and to observe whether the serum concentration of BPA is related to leiomyoma growth, divided leiomyoma patients into three groups; the mean BPA concentrations were 0.274 ± 0.063 ng/mL in the mild group, 0.346 ± 0.064 ng/mL in the moderate and 0.647 ± 0.039 ng/mL in the severe group [34]. Fenvalerate, widely used for its high insecticidal potency and low mammalian toxicity, is classified as an environmental chemical. Data show that data show that Fen can stimulate the growth of both uterine leiomyoma and uterine smooth muscle cells, which involves a combination of enhanced cell cycle progression and inhibition of apoptosis. Also this compound can increase collagen I expression, at both mRNA and protein levels. Interestingly, the ER is less likely involved in either the hyperplasia or extracellular matrix overproduction induced by fenvalerate in human uterine leiomyoma [35].
Dioxins are byproducts of combustion and of multiple industrial processes; TCDD, is the most potent dioxin of the series. In cell culture studies, TCDD, by binding to the aryl hydrocarbon receptor, interacts with estrogen receptors, and thus behaves either as an estrogen agonist or antagonist. Furthermore, in mice, TCDD blocks estrogen-induced responses in several tissues [36]. Possibly, that is why in the Seveso industrial accident, TCDD exposure appeared to reduce the risk of uterine cancer development, still the number of cases was too small for risk assessment of exposure to this xenoestrogen [37,38].
DES was initially used for treatment of postmenopausal symptoms, endometriosis and as an emergency contraceptive, but quickly became an accepted intervention for prevention of miscarriage, premature birth, and other pregnancy problems. Although clinical trials in the 1950s demonstrated that it was ineffective for prevention of adverse pregnancy outcomes, it remained in use until 1971. In 1971, in utero exposure to DES was linked to the occurrence of vaginal clear cell adenocarcinoma in female offspring [39], which prompted the United States Food and Drug Administration (USFDA) to advise against its use in pregnancy [40]. BPA is widely used in the manufacture of polycarbonate plastics, epoxy resins, dental sealants, and as a stabilizing agent in plastics such as polyvinyl chloride [41]. The xenoestrogen BPA increases uterine wet weight, luminal epithelial cell height and the thickness of both the stroma and myometrium in the ovariectomized B6C3F1 mouse. Co-administration of the antiestrogen ICI blocks the increase in uterine wet weight and epithelial cell height, and suggests that these BPA-induced events are mediated by the ER [42]. The ability of BPA to affect human ER binding, expression of PR mRNA and protein, and cell proliferation has been measured in the human endometrial cell line, ECC-1. Although less potent than 17beta-estradiol, BPA was able to bind to the human uterine ER. BPA also induced both mRNA and protein to levels similar to E2 [43]. In human carcinogenesis, the overexpression of cyclooxygenase-2 (COX-2) and epithelial-mesenchymal transition (EMT) are closely related with tumor development. Human endometrial carcinoma cells line (RL95-2) was used to investigate whether BPA can induce EMT and COX-2 expression. The results show that BPA increased growth rate and colony-forming efficiency in a dose-dependent manner, induced EMT and COX-2 gene expression and promoted the migration and invasion ability of RL95-2 cells [44]; probably set here are rare studies demonstrating in vitro the dual role of estrogenicity exerted by BPA in human endometrial cancer.
Obesogenic environment
The next event was the introduction of the term 'obesogen' as representing an environmental pollutant that adversely affects various aspects of adipose tissue functions [45]. The list of cancers at increased risk of development in an 'obesogenic' environment include common adult cancers such as endometrium, post-menopausal breast, colon and kidney, but also less common malignancies such as leukaemia, multiple myeloma, and non-Hodgkin's lymphoma [46]. But, are there common pathophysiologic mechanisms leading to the development of different cancer entities? Insulin resistance is the core mechanism. However, there are several other candidate systems including insulin-like growth factors, sex steroids, adipokines, obesity-related inflammatory markers, the nuclear factor kappa beta (NF-κ B) system and oxidative stresses [46]. Roberts and colleagues brought into the "one system fits all" mechanism three novel candidates: obesityinduced hypoxia, shared genetic susceptibility, and migrating adipose stromal cells [47]. In prospective epidemiological studies, endometrial [48] cancers have been observed in relation to higher levels of circulating C-peptide levels, a biomarker of insulin secretion. In addition, prediagnostic levels of testosterone, androstenedione, dehydroepiandrosterone sulfate, sex hormone-binding globulin, estrone, estradiol, insulin-like growth factor-binding proteins 1 and 2, adiponectin, high- and low-density lipoprotein cholesterol, glucose, triglycerides, tumor necrosis factor (TNF) α, soluble TNF receptors 1 and 2, C-reactive protein, interleukin-6, and interleukin-1 receptor antagonist were measured in 233 incident endometrial cancer cases and 446 matched controls. Factor analysis identified 3 components associated with postmenopausal endometrial cancer risk that could be labeled 'insulin resistance/metabolic syndrome,' 'steroids,' and 'inflammation' factors [48].
It should be mentioned at this point, that indirect evidence for a role of insulin on cancer development is provided by studies showing that type 2 diabetic patients who get insulin therapy or drugs to stimulate insulin secretion have a significantly higher incidence of cancer than those who get metformin, an antidiabetic drug that reduces glucose production in the liver [49,50]. Hyperinsulinaemia could also promote carcinogenesis indirectly by increasing the levels of circulating free IGF-1 that has mitogenic and anti-apoptotic activity [51]. In fact, increased insulin, IGF-1 and IGF-2 levels are associated with tumor growth in vitro, in animal models, and in epidemiological studies in humans [52]. Insulin-like growth factor 1 (IGF-1) is a mitogen which plays a key role in regulating cell proliferation, differentiation, and apoptosis. It belongs to the family of proteins also composed of insulin-like growth factor 2 (IGF-2), two types of membrane receptors (IGF-1R and IGF-2R), 6 binding proteins (IGFBP 1–6), hydrolyzing proteases, and reactive molecules binding proteins, which regulate the activity of growth factors [53]. Cai and colleagues, on attempting to assess the interaction of metformin, insulin-like growth factor 1 (IGF-1) expression, and phosphorylated mammalian target of rapamycin (p-mTOR) and AMP-activated protein kinase (p-AMPK), found that high IGF-1 plasma concentrations in women with endometrial cancer were reversed by conventional antidiabetic doses of metformin [54].
Adiponectin and leptin are the most abundant adipokines. Leptin, abundant in the 'obesogenic environment' has been shown to be mutagenic, pro-inflammatory, anti-apoptotic, and pro-angiogenic; adiponectin is inversely related to BMI and visceral adiposity, may stimulate apoptosis and inhibit angiogenesis and cell migration. In prospective studies, adipokines have been found to be associated with the risk of cancer of endometrium [55]. Leptin enhances the production of inflammatory factors, and different pathways link inflammation and cancer by a number of oncogenes [56]. In fact, chemokines are key players of the cancer-related inflammation− components of an intensive dialogue promoting angiogenesis, metastasis, subversion of adaptive immunity and changing response to hormones and to chemotherapeutic agents [57,58]. Emerging evidence also suggests that cancerrelated inflammation promotes genetic instability [58], reinforcing the suggestion of shared and modified genetic susceptibility to cancer of the endometrium. Several genes have been found to play an important role in the etiology of obesity and tumourigenesis. For instance, FTO gene polymorphism may be a potential biomarker in early diagnosis or gene therapy target of endometrial cancer and pancreatic cancer [59].
Epilogue and Future Goals
Although there are many uncertainties about basic mechanisms in the control of cell proliferation and carcinogenesis at the present time (dose, time and route of exposure), the question of whether xenoestrogens are a risk factor for endometrial cancer has to be addressed by measuring the estrogen burden of these chemicals in human blood.
Conclusion
To conclude, a large research program is needed to find out whether or not current levels of xenoestrogen exposure increase the incidence of hormone-related endometrial neoplasia over mechanisms that have been extensively described in the literature.
18106
References
- Bergeron JM, Crews D, McLachlan JA (1994) PCBs as environmental estrogens: Turtle sex determination as a biomarker of environmental contamination. Environ Health Perspect 102: 780-781.
- Sager DB, Shih-Scaroeder W, Girand D (1987) Effect of early postnatal exposure to polychlorinated biphenyls (PCBs) on fertility in male rats. Bull Environ Contam Toxicol 38: 946-953.
- Dieringer CS, Lamartiniere CA, Schiller CM, Lucier GW (1979) Altered ontogeny of hepatic steroid-metabolizing enzymes by pure polychlorinated biphenyl congeners. Biochem Pharmacol 28: 2511-2514.
- Woodward AR, Percival HF, Jennings ML, Moore CT (1993) Low clutch viability of American alligators on Lake Apopka. Fla Sci 56: 52-63.
- Guillette LJ, Gross TS, Masson GR, Matter JM, Percival F, et al. (1994) Developmental abnormalities of the gonad and abnormal sex hormone concentrations in juvenile alligators from contaminated and control lakes in Florida. Environ Health Perspect 102: 681-688.
- Feuer EJ, Wun LM (1992) How much of the recent rise in breast cancer can be explained by increases in mammography utilization? Am J Epidemiol 136: 1423-1436.
- Henderson BE, Ross RK, Pike MC (1993) Hormonal chemoprevention of cancer in women. Science 259: 633-638.
- Wolff MS, Paolo G, Toniolo P, Lee EW, Rivera M, et al. (1993) Blood levels of organochlorine residues and risk of breast cancer. J Natl Cancer Inst 85: 648-652.
- Davis DL, Bradlow HL, Wolff M, Woodruff T, Hoel DG, et al. (1993) Medical hypothesis: Xenoestrogens are preventable causes of breast cancer. Environ Health Perspect 101: 372-377.
- Wheeler JM (1992) Epidemiology and prevalence of endometriosis. Infertil Reprod Med Clin 3: 545-549.
- Rier SE, Martin DC, Bowman RE, Dmowski WP, Becker L, et al. (1993) Endometriosis in rhesus monkeys (Macaca mulatta) following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fund Appl Toxicol 21: 433-441.
- Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R (1991) 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds as antiestrogens: Characterization and mechanism of action. Pharmacol Toxicol 69: 400-409.
- Bois FY, Eskenazi B (1994) Possible risk of endometriosis for Seveso, Italy, residents: An assessment of exposure to dioxin. Environ Health Perspect 102: 476-477.
- Uwing J (1916) Pathological aspects of some problems of experimental cancer Res J Cancer Res: 71-86.
- Liehr JG, Stancel GM, Chorich LP, Bousfield GR, Ulubelen AA (1986) Hormonal carcinogenesis: Separation of estrogenicity from carcinogenicity. Chem Biol Interactions 59: 173-184.
- Roy D, Bernhardt A, Strobel HW, Liehr JG (1992) Catalysis of the oxidation of steroid and stilbene estrogens to estrogen quinine meatabolites by the beta-naphthoflavone-inducible cytochrome P450IA family. Arch Biochem Biophys 296: 450-456.
- Soto AM, Lin TM, Sakabe K, Olea N, Damassa DA, et al. (1995) α-Variants of the human prostate LNCaP cell line as a tool to study discrete components of the androgen-mediated proliferative response. Oncol Res 7: 545-558.
- Szelei J, Jimenez J, Soto AM, Luizzi MF, Sonnenschein C (1997) Androgen induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology 138: 1406-1412.
- Shaw E, Farris M, McNeil J, Friedenreich C (2016) Obesity and endometrial cancer. Recent Results. Cancer Res 208: 107-136.
- Bokhman JV (1983) Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 15: 10–17.
- Sherman ME, Bur ME, Kurman RJ (1995) p53 in endometrial cancer and its putative precursors: evidence for diverse pathways of tumorigenesis. Hum Pathol 26: 1268–1274.
- Hale GE, Hughes CL, Cline JM (2002) Endometrial cancer: hormonal factors, the perimenopausal "window of risk," and isoflavones. J Clin Endocrinol Metab 87: 3-15.
- Misichronis G, Georgopoulos NA, Marioli DJ, Armeni AK, Katsikis I, et al. (2011) The influence of obesity on Androstenedione to Testosterone ratio in women with polycystic ovary syndrome (PCOS) and hyperandrogenemia. Gynecol Endocrinol.
- Visvanathan K, Vang R, Shaw P, Gross A, Soslow R, et al. (2011) Diagnosis of serous tubal intraepithelial carcinoma based on morphologic and immunohistochemical features: a reproducibility study. Am J Surg Pathol 35: 1766-1775.
- Newbold RR, Jefferson WN, Padilla-Banks E (2007) Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol 24: 253-258.
- Couse JF, Davis VL, Hanson RB, Jefferson WN, McLachlan JA, et al. (1997) Accelerated onset of uterine tumors in transgenic mice with aberrant expression of the estrogen receptor after neonatal exposure to diethylstilbestrol. Mol Carcinog 19: 236-242.
- Couse JF, Korach KS (2004) Estrogen receptor-alpha mediates the detrimental effects of neonatal diethylstilbestrol (DES) exposure in the murine reproductive tract. Toxicology 205: 55-63.
- Jefferson WN, Chevalier DM, Phelps JY, Cantor AM, Padilla-Banks E, et al. (2013) Persistently altered epigenetic marks in the mouse uterus after neonatal estrogen exposure. Mol Endocrinol 27: 1666-1677.
- Louis GW, Hallinger DR, Stoker TE (2013) The effect of triclosan on the uterotrophic response to extended doses of ethinyl estradiol in the weanling rat. Reprod Toxicol 36: 71-77.
- Yoshida M, Inoue K, Takahashi M (2015) Predictive modes of action of pesticides in uterine adenocarcinoma development in rats. J Toxicol Pathol 28: 207-216.
- Katz TA, Yang Q, Treviño LS, Walker CL, Al-Hendy A (2016) Endocrine-disrupting chemicals and uterine fibroids. Fertil Steril 106: 967-977.
- Jeong EH, Hong GY, Kim BR, Park SN, Lee HH, et al. (2013) The relationship between uterine myoma growth and the endocrine disruptor in postmenopausal women. J Menopausal Med 19: 130-134.
- Gao X, Yu L, Castro L, Moore AB, Hermon T, et al. (2010) An endocrine-disrupting chemical, fenvalerate, induces cell cycle progression and collagen type I expression in human uterine leiomyoma and myometrial cells. Toxicol Lett 196: 133-141.
- Buchanan DL, Sato T, Peterson RE, Cooke PS (2000) Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in mouse uterus: critical role of the aryl hydrocarbon receptor in stromal tissue. Toxicol Sci 57: 302-311.
- Landi MT, Consonni D, Patterson DG Jr, Needham LL, Lucier G, et al. (1998) 2,3,7,8-Tetrachlorodibenzo-p-dioxin plasma levels in Seveso 20 years after the accident. Environ Health Perspect 106: 273-277.
- Eskenazi B, Warner M, Samuels S, Young J, Gerthoux PM, et al. (2007) Serum dioxin concentrations and risk of uterine leiomyoma in the Seveso Women's Health Study. Am J Epidemiol 166: 79-87.
- Herbst AL, Ulfelder H, Poskanzer DC (1971) Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med 284: 878.
- FDA Drug Bulletin (1971) Diethylstilbestrol contraindicated in pregnancy. US Department of Health, Education, and Welfare. Washington, DC.
- Staples CA, Dorn PB, Klecka GM, O'Block ST, Harris LR (1998) A review of the environmental fate, effects, and exposures of bisphenol A. Chemosphere 36: 2149–2173.
- Papaconstantinou AD, Umbreit TH, Fisher BR, Goering PL, Lappas NT, et al. (2000) Bisphenol a-induced increase in uterine weight and alterations in uterine morphology in ovariectomized B6C3F1 mice: Role of the estrogen receptor. Toxicol Sci 56: 332-339.
- Bergeron RM, Thompson TB, Leonard LS, Pluta L, Gaido KW (1999) Estrogenicity of bisphenol-A in a human endometrial carcinoma cell line. Mol Cell Endocrinol 150: 179-187.
- Wang KH, Kao AP, Chang CC, Lin TC, Kuo TC (2015) Bisphenol A-induced epithelial to mesenchymal transition is mediated by cyclooxygenase-2 up-regulation in human endometrial carcinoma cells. Reprod Toxicol 58: 229-233.
- Decherf S, Demeneix BA (2011) The obesogen hypothesis: a shift of focus from the periphery to the hypothalamus. J Toxicol Environ Health B Crit Rev 14: 423-448.
- Renehan AG, Roberts DL, Dive C (2008) Obesity and cancer: Pathophysiological and biological mechanisms. Arch Physiol Biochem 114: 71-83.
- Roberts DL, Dive C, Renehan AG (2010) Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med 61: 301-316.
- Dossus L, Lukanova A, Rinaldi S, Allen N, Cust AE, et al. (2013) Hormonal, metabolic, and inflammatory profiles and endometrial cancer risk within the EPIC cohort--a factor analysis. Am J Epidemiol 177: 787-799.
- Chlebowski RT, McTiernan A, Wactawski-Wende J, Manson JE, Aragaki AK, et al. (2012) Diabetes, metformin, and breast cancer in postmenopausal women. J Clin Oncol 30: 2844-2852.
- Thakkar B, Aronis KN, Vamvini MT, Shields K, Mantzoros CS (2013) Metformin and sulfonylureas in relation to cancer risk in type II diabetes patients: a meta-analysis using primary data of published studies. Metabolism 62: 922-934.
- Gallagher EJ, LeRoith D (2011) Mini review: IGF, Insulin, and Cancer. Endocrinology 152: 2546-2551.
- Gallagher EJ, LeRoith D (2010) The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol 3etab 21: 610-618.
- Majchrzak-Baczmańska D, Malinowski A (2016) Does IGF-1 play a role in the biology of endometrial cancer? Ginekol Pol 87: 598-604.
- Cai D, Sun H, Qi Y, Zhao X, Feng M, et al. (2016) Insulin-like growth factor 1/mammalian target of rapamycin and AMP-activated protein kinase signaling involved in the effects of metformin in the human endometrial cancer. Int J Gynecol Cancer 26: 1667-1672.
- Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS (2005) Low plasma adiponectin levels and risk of colorectal cancer in men: A prospective study. J Natl Cancer Inst 97: 1688-1694.
- Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454: 436-444.
- Germano G, Allavena P, Mantovani A (2008) Cytokines as a key component of cancer-related inflammation. Cytokine 43: 374-379.
- Mantovani A, Garlanda C, Allavena P (2010) Molecular pathways and targets in cancer-related inflammation. Ann Med 42: 161-170.
- Huang X, Zhao J, Yang M, Li M, Zheng J (2016) Association between FTO gene polymorphism (rs9939609 T/A) and cancer risk: a meta-analysis. Eur J Cancer Care (Engl).