Keywords
PET/CT; Basal ganglia; 18F-FDG
Introduction
Wilson disease (WD) also known as hepatolenticular degeneration) is one of the 42 hereditary disorders related to dystonic symptoms [1]. First described in 1912 by Kinnear Wilson [2], it is an autosomal-recessive disorder caused by mutation in the ATP-gene, with copper accumulation, first in the liver, brain and other tissues, with clinical manifestations that may include hepatic, neurological, psychiatric and ophtalmological derangements. It has an average prevalence of 30/1,000,000. The age at which WD may present or be diagnosed is between 5 and 35 years of age. [3].
The disturbance of copper metabolism in WD was described in 1948 by Mendelbrote and Cunnings [4]. Copper is an essential metal that is an important cofactor for many proteins. It is absorbed by enterocytes mainly in the duodenum and proximal small intestine and transported in the portal circulation in association with albumin and the amino acid histidine to the liver, where it is avidly removed from the circulation. The liver utilizes some of this metal for metabolic needs, synthesizes and secretes the copper-containing protein ceruloplasmin, and excretes excess copper into bile. Processes that impair biliary copper excretion can lead to increases in hepatic copper content. A mutation (300 types) of ATP7B gene, located in chromosome 13, leads to the absent or reduce function of a metal-transporting ATPase, causing a decrease in the hepatocellular excretion of copper into bile, with the subsequent accumulation of this metal and its deposition in different organs with direct toxicity and cellular damage, producing the symptoms of Wilson disease. An additional consequence is the decreased blood level of ceruloplasmin.
Hepatic dysfunction is the initial clinical manifestation in 40 to 50% of individuals with Wilson disease. Kayser-Fleischer rings (deposition of copper in the Decemet membrane, visible as a band of golden-brownish pigment near limbus) is present only in 44-62% of patients with mainly hepatic disease at the time of diagnosis, and in the 95% of patients with neurological presentation, but even in these cases may be absent [5]. Neurological dysfunction constitutes the initial clinical manifestation in 40– 60% of individuals with Wilson disease. Tremor, lack of motor coordination, dystonia, spasticity, dysarthria, dystonia involving the tongue, face, and pharynx may produce not only dysarthria, but also drooling and an unusual perturbation of facial expression that results in a frozen grimace (risus sardonicus). Seizures are an infrequent component of Wilson disease, but may occur in up to 6% of patients. Mutation analysis by whole-gene sequencing is possible and should be performed on individuals in whom the diagnosis is difficult to establish by clinical and biochemical testing [6]. Other findings apart from neurologic or psychiatric disease are renal abnormalities including aminoaciduria and nephrolithiasis, skeletal abnormalities such as premature osteoporosis and arthritis, cardiomyopathy, pancreatitis, hypoparathyroidism, and infertility or repeated miscarriages.
Available treatments include D-penicillamine, trientine, zinc and dietary restrictions.
Secondary dystonia is a large and diverse group of disorders. Of particular interest are the heredo-degenerative diseases, including the metabolic disorders, which are characterized by pathological abnormalities that involve the basal ganglia [7]. The dystonia-parkinsonism syndrome is a combination of akineticrigid and hyperkinetic disorders, sometimes accompanied by pyramidal tract involvement and other neurological deficits. Among the major diseases that can present this syndrome are dopa-responsive dystonia (DRD), Wilson disease, Parkin- PINK1 and DJ-1- associated parkinsonism, X-linked dystoniaparkinsonism/ Lubag (DYT3), rapid-onset dystonia-parkinsonism (DYT12) and neurodegeneration with brain iron accumulation.
Establishing the correct diagnosis can be difficult. The evaluation of dystonia should include an exhaustive medical history, family history and a clinical examination to discard secondary causes. Wilson disease should be ruled out by measuring serum ceruloplasmin levels and 24-hour urinary copper levels and by slit-lamp examination. Dopa-responsive dystonia may be ruled out with a three week trial of levodopa. in vivo imaging, such as 18F-Fluoro-desoxyglucose-Positron Emission-Tomography (18FFDG PET) and functional magnetic resonance imaging (fMRI), provides means to monitor structural and functional changes in the brain of these patients.
In this report, we present the case of a patient with generalized dystonia caused by Wilson disease, the diagnostic process and the contribution of functional imaging with 18F-FDG PET/CT.
Case Presentation
A 30 year old female patient from the state of Queretaro, mother of two children, secondary schooling, and occasional smoker with no family history of abnormal movements; arrived at the Emergency Department with a diagnosis of involuntary movements (CIE-10 R258). She began a year before her arrival to our Institution presenting a shuffling gait with poor arm swing and stooped posture which was progressing throughout the year with the appearance of disabling fixed positions on arms, legs and feet, opisthotonos and lingual and guttural dysarthria that impared communication. The patient also presented drooling and difficulty feeding with weight loss leading to cachexia. On the physical examination, spastic muscle tone with abnormal posture and bilateral extensor plantar response were noted. Cerebellar tests without alterations.
Presumption diagnosis was generalized dystonia-parkinsonism of early adulthood, progressive and persistent, combined with piramidal tract involvement; with topographic localization in the basal ganglia.
The trial of carbidopa-levodopa (one 25/100 mg tablet three times a day) ruled out dopa-responsive dystonia. Normal antibodies discarded an autoimmune disease. An electroencephalogram ruled out epilepsy. The slit-lamp examination performed by a neuro-ophtalmologist was normal. The penicillamine challenge test was not available at the moment. The echocardiogram showed hypertrophic cardiomyopathy. The MRI did not show structural abnormalities of the basal ganglia.
PET brain study was performed on a Siemens Biograph 64 mCT PET/CT (Siemens Medical Solutions) according to the standard protocol (procedure guideline for FDG PET Brain imaging). The patient fasted 6 hrs prior to intravenous injection of 18F-FDG (3.7MBq/kg body weight). Blood glucose level was 81 mg/ dl. After a 30 min uptake period, during which the patient was instructed to rest silently, static imaging were acquired.
Brain 18F-FDG PET/CT (Figure 1) showed a severe bilateral decrease uptake of the tracer at the corpus striatum; thalamus uptake was preserved. The rest of the study was normal. A whole body scan highlighted a pleural and pericardial effusion and fluid ascites (Figure 2). Low serum ceruloplasmin levels (10.4 UI/ mL), elevation of aminotransferase activity and 24-hour urinary copper levels, associated with PET results, led to the diagnosis of Wilson disease.
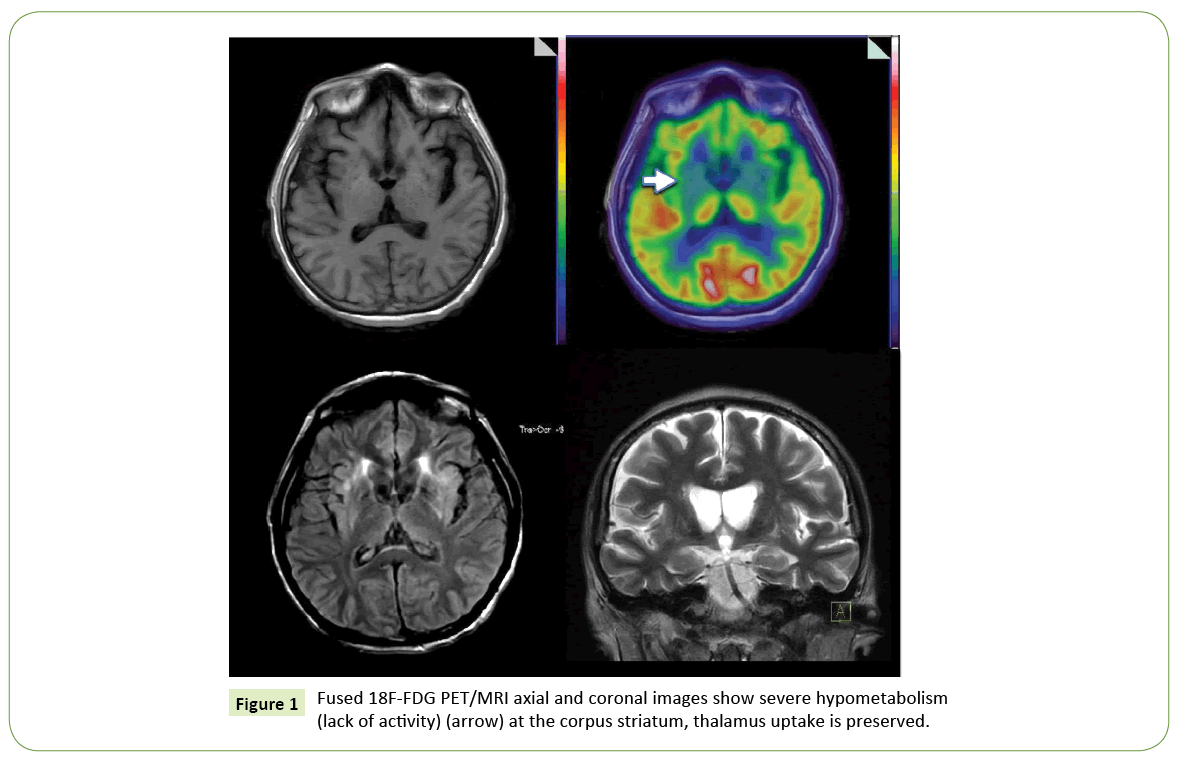
Figure 1: Fused 18F-FDG PET/MRI axial and coronal images show severe hypometabolism (lack of activity) (arrow) at the corpus striatum, thalamus uptake is preserved.
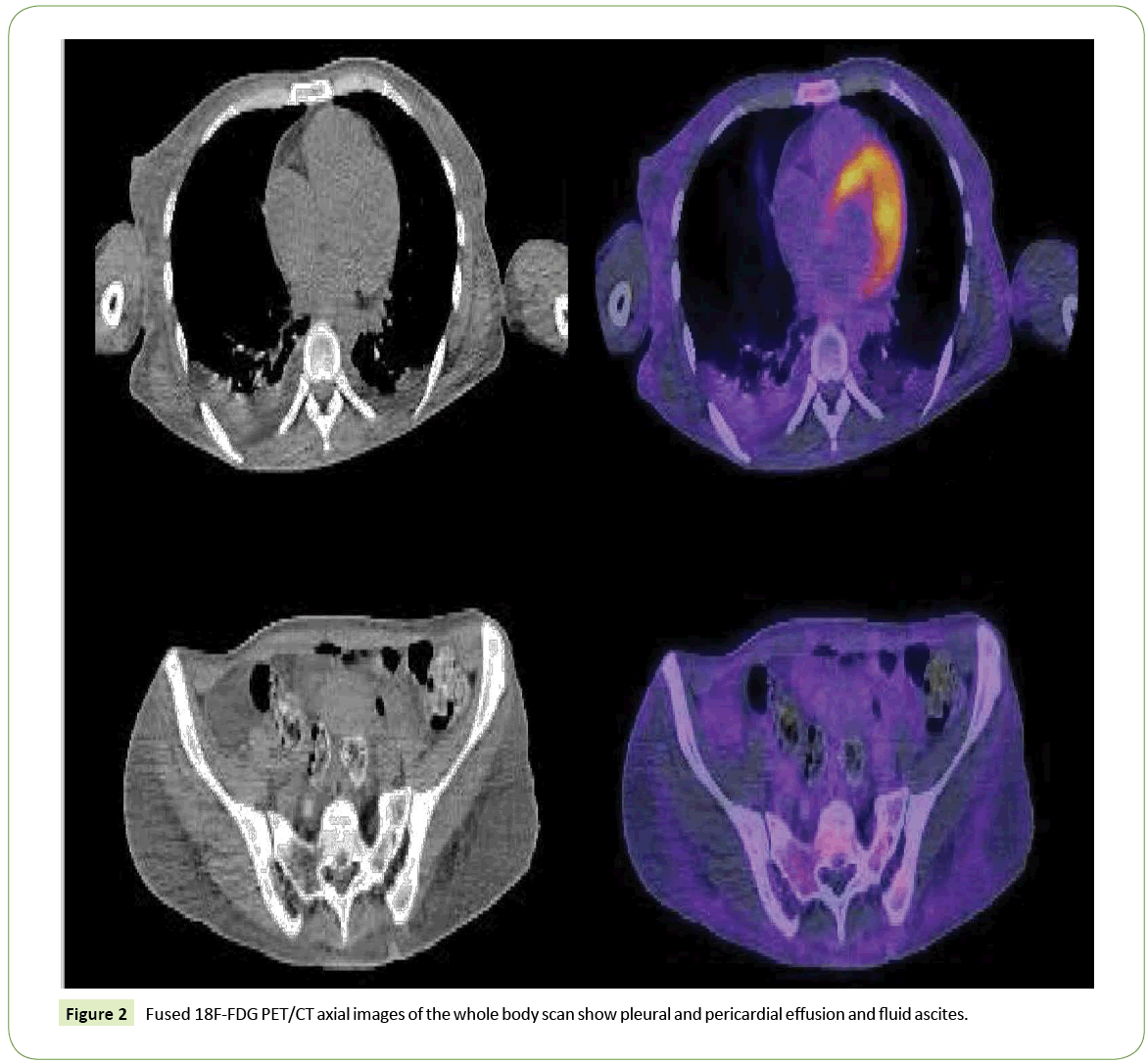
Figure 2: Fused Figure 2 18F-FDG PET/CT axial images of the whole body scan show pleural and pericardial effusion and fluid ascites.
Discussion
The basal ganglia are a group of nuclei situated in the deep gray matter at the base of the fore-brain that are interconnected with the cerebral cortex, thalami and brainstem. In a strict anatomical sense they contain three paired nuclei: caudate nucleus, putamen (together with the caudate nucleus known as corpus striatum) and globus pallidus (together with the putamen known as the lentiform nucleus) within each cerebral hemisphere. Functionally, two additional nuclei are also part of the basal ganglia, the subthalamic nuclei and the substantia nigra. The primary contributors to the basal ganglia vascular supply are the medial and lateral lenticulostriate arteries, which arise from the anterior and middle cerebral arteries, respectively; and the recurrent artery of Heubner. Venous drainage of both the basal ganglia and the thalamus is into the deep venous system.
The basal ganglia are highly metabolically active, rich in mitochondria, vascular supply and neurotransmitters (GABA, dopamine, acetylcholine and glutamine).
The functions of the basal ganglia are complex. They are mainly involved in the production of movement and are a part of the extrapyramidal motor system, and they also contribute to higher emotional, sensorimotor, associative and cognitive functions.
These characteristic features of the basal ganglia make them susceptible to injury (toxic poisoning, metabolic abnormalities and neurodegeneration with brain iron accumulation), producing symmetric affection.
The suggestion that dystonia primarily arise from a disorder of basal ganglia function is known since the 80s, and it is based on an imbalance between the direct excitatory and indirect inhibitory output pathways. The striatum is the input region of the basal ganglia, and it receives corticostriatal fibers as well as the dopaminergic nigrostriatal projection. Neuroimaging has provided strong evidence that corroborate this knowledge [8].
Neuroimaging offers a non-invasive method to examine structural and functional changes in the evaluation of those pathologies. The magnetic resonance (MR) imaging is the modality of choice for evaluating the basal ganglia, but computed tomography (CT) may be useful as the first line of the medical approach [9]. On axial brain images, the caudate nucleus is C-shaped, and the putamen a curved structure bordered on its convex margin by the external capsule. The lentiform nucleus and the head of the caudate nucleus can be visualized as paired symmetric structures located between the lateral ventricle and the insular cortex, and it is separated from the caudate head and the thalamus by the anterior and posterior limbs of the internal capsule, respectively. The radiographic appearance of basal ganglia is normally isodense/isointense with cortex [10,11].
Neuroimaging of the basal ganglia in Wilson disease
In Wilson disease, MRI findings include symmetric putaminal T2 signal-intensity (T2 prolongation), also affecting globus pallidus, caudate nuclei and thalamus. The cortical and subcortical regions, mesencephalon, pons, vermis, and dentate nuclei may also be involved. Diffusion restriction is often seen in the early stages of the disease. T1 may also show hyperintensity within the putamen [12].
PET provides a means of studying regional cerebral function in vivo under resting and activating conditions. Studies on blood flow and metabolism in dystonia have tended to yield conflicting results, with reports varying from normal, increased and decreased resting striatal metabolism [13]. Little has been written about 18F-FDG PET/CT for Wilson disease. However, studies with small samples of patients with this diagnosis, describe a decrease in the regional cerebral glucose metabolic rate in caudate and lentiform nucleus, with preservation of the thalamus [14,15]. Global cerebral glucose metabolism is unaltered [16].
Numerous molecular imaging studies with PET or single-photon emission computed tomography (SPECT) that investigate dopamine receptors and endogenous dopamine release demonstrate the effectiveness of investigating the dopaminergic pathways for this pathology [17]. Different investigations of dopamine receptors with PET/CT using [123 I]2- carbomethoxy- 3-(4[123I]iodophenyl)tropane ([123I]- CIT), showed that the integrity of the nigrostriatal dopaminergic neurons in the striatum of patients with WD is differentially altered depending on the course and severity of the disease [18,19].
Basal Ganglia evaluation with 18F-FDG PET/CT
The brain consumes 20% of the body’s energy. In 1977, Sokoloff showed the correlation between brain function and increased blood flow and glucose utilization [20]. In recent years, it has been demonstrated that the interaction between astroglial cells and neuronal cells, by glutamate-mediated signaling and a Ca2+- dependent astrocytic mechanism, produces an increase in the cerebral blood flow to an extent that more oxygen and glucose is provided to active brain regions than is consumed [21].
18F-Fluorodeoxyglucose (FDG) is an index molecule for glucose metabolism. Positron emission tomography using this tracer allows the evaluation of changes in regional cerebral metabolism. Glucose is the principal metabolic substrate for the brain cells, which provides approximately 95% of adenosine triphosphate (ATP) required for brain function. There, it is almost entirely oxidized through sequential glycolysis and the tricarboxylic cycle associated with oxidative phosphorylation; and can also be stored as glycogen. Brain gray matter is responsible for the massive energy consumption [33 to 50 μmol ATP/g/min in neocortex) [22], most of it serves to restore the membrane gradient following neuronal depolarization, neurotransmitter recycling, intracellular signaling and dendritic and axonal transport. Glial and ependymal cells also have their own metabolic needs.
The properties of the neurons within the basal ganglia resemble the characteristics of neurons within the cortical areas that form the loops that control the different motor and cognitive behavioral functions. Various studies using functional imaging had demonstrated the increased irrigation and glucose consumption to these nuclei in relation with the aforementioned functions [23]. According to this data, it can be expected that the alteration of functional activity in the basal ganglia will be associated with changes in regional cerebral metabolism (rCMR) and regional blood flow (rCBF). This has been confirmed for Parkinson [24] and Huntington's disease [25], among others [26], and provides a means for assessing Wilson disease under such premises.
Conclusion
As shown in this case presentation, the 18F-FDG PET/CT is useful for the recognition of Wilson disease when there is a high clinical suspicion, with the advantage of performing at the same time the identification of extra cranial manifestations. The integration of nuclear neurosciences in addressing patients is essential for a proper and timely diagnosis.
20769
References
- Breakfield XO, Blood AJ, Li Y (2008)The pathophysiological basis of dystonias, Nature Reviews 9: 222-234.
- Wilson SAK (1912)Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 34:295-507.
- Cumings JN (1948)The copper and iron content of brain and liver in the normal and inhepato-lenticular degeneration. Brain 71:410–441.
- Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, et al. (1997) Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 113:212-218.
- Roberts EA, Schilsky ML (2008)Diagnosis and treatment of Wilson Disease: An Update. Hepatology 47: 2089-2111.
- Toessi J, Lehericy S, Strafella AP (2014)Imaging insights into basal ganglia function, Parjinson´s disease and dystonia. Lancet 384: 532-544.
- Hedge AN, Mohan S, Lath N, Lim CCT (2011) Differential diagnosis for bilateral abnormalities of the basal ganglia and thalamus. RadioGraphics 31: 5-30.
- Telford R, Vattoth S (2014)MR Anatomy of deep brain nuclei with special reference to specific diseases and deep brain stimulation localization. The Neuroradiology Journal 27:29-43.
- Lincoln CM, Bello JA, Lui YW (2012)Decoding the deep gray: A review of the anatomy, function, and imaging patterns affecting the basal ganglia.Neurographics 2: 92-102.
- Brooks DJ (1995)The role of the basal ganglia in motor control: contributions from PET, J NeurolSci 128:1-13.
- Hawkins RA, Mazziotta JC, Phelps ME (1987)Wilson´s disease studied with FDG and positron emission tomography. Neurology 37:1707.
- Karbe H, Holhoff VA, Rudolf J (1992)Positron emission tomography demonstrates frontal cortex and basal ganglia hypometabolism in dystonia. Neurology 42: 1526-1563.
- Kumar S, Prabhakar S (2006)Positron emission tomography in neurological diseases, Neurology India 53: 149-155.
- Iacoboni M, Baron JC, Frackowiak SJ (1999)Emission tomography contribution to clinical neurology. Clinical Neurophysiology 110:2-23.
- Karimi M, Perimutter JS (2015)The role of dopamine and dopaminergic pathways in Dystonia: Insights from Neuroimaging. Tremor Other HyperkinetMov 5: 1-10.
- Barthel H, Hermann W, Kluge R (2003)Concordant Pre- and postsynaptic deficits of dopaminergic neurotransmission in neurologic Wilson disease. AJNR Am J Neuroradiol 24: 234-238.
- Jeon B, Kim JM, Jeong JM (1998)Dopamine transporter imaging with [123I]-B-CIT demostrataes presynaptic nigrostriatal dopaminergic damage in Wilson´s disease. J NeurolNeurosurgPsichiatry 56:60-64.
- Sokoloff L, Reivich M, Kennedy C (1977)The 14C-deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure and normal values in the conscious and anesthetized albino rat. J Neurochem 28: 897-916.
- Atwell D, Buchan AM, Charpak S (2010) Glial and neuronal control of brain blood flow. Nature 468: 231-243.
- MacVicar BA, Newman EA (2015) Astrocyte regulation of blood flow in the brain. Cold Spring HarbPerspectBiol 1-14.
- Atwell D, Laughlin SB (2001)Review Article: An energy budget for signaling in the grey matter of the brain, Journal of Cerebral Blood Flow and Metabolism21: 1133-1145.
- Hsu JL, Jung TP, Hsu CY (2007)Regional CBF changes in Parkinson’s disease: a correlation with motor dysfunction, Eur J Nucl Med Mol Imaging 34:1458-1466.
- Chen JJ, Salat DH, Rosas D (2012)Complex relationships between cerebral blood flow and brain atrophy in early Huntington’s disease, Neuroimage 59: 1043-1051.
- Leisman G, Melillo R, Carrick FR (2012)Clinical motor and cognitive neurobehavioral relationships in the basal ganglia, InTechpp: 1-30.

