Keywords
Parkinson; Pharmacogenetics; Dopaminergic therapy; Side effects
Introducción
Farmacogenética y farmacogenómica
Una reacción adversa a medicamentos (RAM) es cualquier respuesta a un medicamento que sea nociva y no intencionada, y que tenga lugar a dosis que se apliquen normalmente en el ser humano. Las RAM causan más de 2 millones de hospitalizaciones y 100000 muertes al año en Estados Unidos. La incidencia de RAM graves en pacientes hospitalizados es de entre el 6% y el 10%, con un desenlace fatal en aproximadamente el 0,32% [1,2].
Las RAM pueden ser debidas múltiples determinantes, ambientales o genéticos. Para tratar de comprender la susceptibilidad interindividual a las consecuencias clínicas de los fármacos se están desarrollando dos campos de conocimiento de rápido crecimiento: La farmacogenética (investigación centrada en uno o unos pocos genes) y la farmacogenómica (investigación centrada en muchos genes al mismo tiempo). La secuenciación del genoma humano [3] ha puesto de manifiesto que los polimorfismos de nucleótidos simples (SNPs) suponen la mayor fuente de variación entre individuos, representando más del 90% de todo el polimorfismo de DNA. Un SNP es una variación en la secuencia de ADN en la que un solo nucleótido (A, T, C o G) en el genoma difiere entre los pares de cromosomas en un individuo. Una variación genética puede ser considerada como “SNP” cuando aparece con una frecuencia igual o superior al 1% en la población. Éste y otros polimorfismos genéticos, tales como las repeticiones variables en tándem (VNTRs) y las inserciones y deleciones (ins/dels) pueden afectar el metabolismo de fármacos, la absorción, la distribución y la eliminación, influenciando en último término la eficacia y los efectos adversos, y las diferencias interindividuales. Por tanto, la identificación y caracterización de polimorfismos genéticos (biomarcadores genéticos) correspondientes a genes que codifican para enzimas metabolizadoras o proteínas transportadoras de fármacos puede proporcionar un conocimiento sustancial sobre los mecanismos subyacentes a las diferencias interindividuales en la respuestas a fármacos, permitiendo optimizar la estrategia terapéutica de cada paciente para maximizar la eficacia y minimizar las RAM mediante una prescripción personalizada al perfil genético de cada paciente.
Enfermedad de parkinson
La Enfermedad de Parkinson (EP) es un trastorno del movimiento progresivo caracterizado por temblor, rigidez, bradicinesia e inestabilidad postural. La EP tiene una prevalencia aproximada de 0.5-1% en personas de 65 a 69 años, elevándose hasta el 1-3% entre los mayores de 80 años [4]. Tan sólo el 5-10% de los sujetos tienen una enfermedad de origen genético con herencia Mendeliana clásica [5]; la mayor parte de los casos son EP esporádica, en los que se asume que la enfermedad se produce por interacción de múltiples factores genéticos y ambientales aún no bien conocidos. El marcador neuropatológico principal de la enfermedad es la pérdida de neuronas dopaminérgicas en la sustancia negra y otras regiones cerebrales y el acúmulo de una proteína, denominada alfa-sinucleína, que forma depósitos conocidos conocidos como Cuerpos de Lewy.
La Levodopa es un precursor de la dopamina que se absorbe en el tracto gastrointestinal y atraviesa la barrera hematoencefálica (Figura 1). La levodopa (L-Dopa) ha demostrado ser el mejor tratamiento sintomático para la EP. De todos modos, su eficacia a largo plazo es limitada por complicaciones relacionadas con el uso sostenido de la medicación, tales como discinesias y fluctuaciones motoras. Cuando se administra por vía oral se metaboliza a nivel periférico por la enzima decarboxilasa (I-AAADC) y la COMT, obteniendo metabolitos activos que causan efectos secundarios a nivel sistémico. La L-Dopa suele administrarse combinada con un inhibidor periférico de la dopa-decarboxilasa para evitar la conversión de L-Dopa a Dopamina fuera del cerebro, lo que permite aumentar su biodisponibilidad a nivel central ya que la L-Dopa puede atravesar la barrera hematoencefálica mientras que la Dopamina no (Figura 1). Los agonistas dopaminérgicos, los inhibidores de la catecol-o-metiltransferasa (COMT) y los inhibidores de la monoaminoxidasa B (IMAO-B) son otros fármacos que han demostrado utilidad en el tratamiento de la enfermedad (Figura 2), presentando también su propio perfil de efectos secundarios. Aunque en fases iniciales de la enfermedad los IMAO y los agonistas dopaminérgicos se pueden utilizar sin necesidad de combinarlos con la L-Dopa, lo habitual es que los pacientes terminen precisando una combinación variable de estos fármacos. De todos modos, como la EP es una enfermedad progresiva, todas las medicaciones terminan resultando insuficientes, por lo que es frecuente la aparición de efectos adversos con el progresivo aumento de las dosis de los fármacos [6,7].
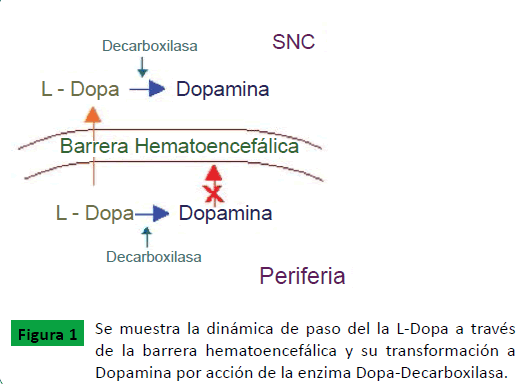
Figura 1: Se muestra la dinámica de paso del la L-Dopa a través de la barrera hematoencefálica y su transformación a Dopamina por acción de la enzima Dopa-Decarboxilasa.
Sin embargo, se ha visto que existe gran heterogeneidad en el tipo e intensidad de RAM que presenta cada paciente. Por ejemplo, hasta el 45% de los pacientes que toman L-Dopa desarrollan discinesias en menos de 5 años, mientras que otros permanecen libres de las mismas durante muchos años más. De igual modo, hasta el 25% de los pacientes que toman agonistas dopaminérgicos presentan alucinaciones, mientras que el 75% de los pacientes, tomando dosis equivalentes, no presentan [8]. Igualmente, la eficacia a los distintos fármacos también es variable, pudiendo unos pacientes responder mejor a la L-Dopa, y otros a los agonistas dopaminérgicos.
También existen fármacos liberadores presinápticos de dopamina (Figura 2), como la Amantadina, aunque su uso no está muy extendido hoy en día porque su eficacia es limitada, quedando prácticamente restringida a los casos con discinesias por levodopa. La Amantadina es un fármaco antiviral con eficacia sintomática limitada sobre la EP. Actúa bloqueando los receptores de la NMDA (N-metil-D-aspartato), aminoácido excitatorio que al encontrarse en concentraciones desequilibradas que provoca una neurodegeneración, favoreciendo la aparición de la EP.
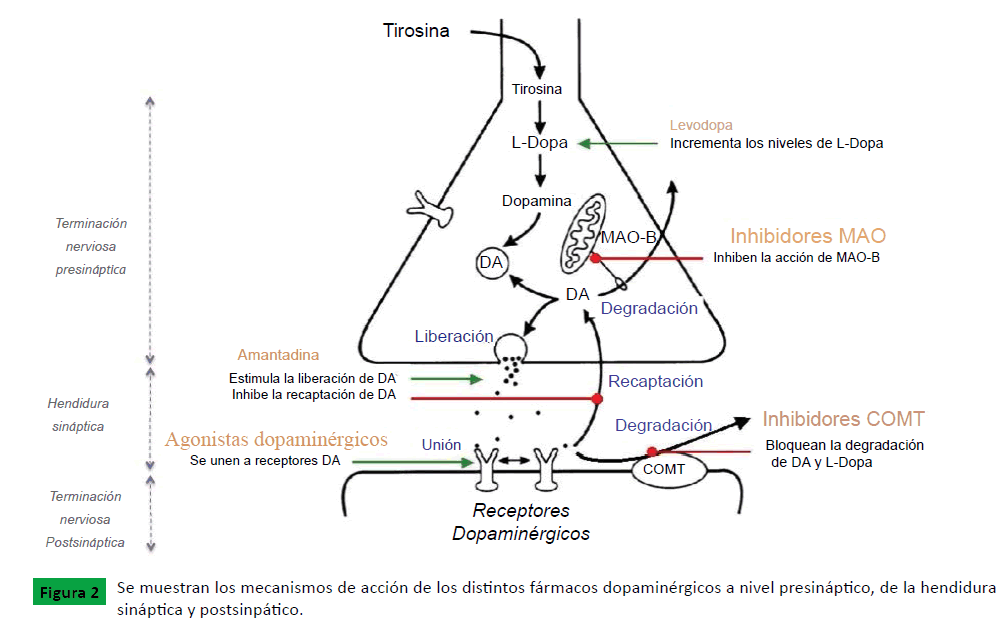
Figura 2: Se muestran los mecanismos de acción de los distintos fármacos dopaminérgicos a nivel presináptico, de la hendidura sináptica y postsinpático.
Aunque generalmente es bien tolerada no está exenta de efectos adversos como cambios en el humor, mareos, retención urinaria, ataxia, xerostomía, confusión y alucinaciones. Por último, los anticolinérgicos de acción central como el Trihexifenidilo, que también pueden aportar cierto beneficio a los pacientes con EP, han caído en desuso por sus frecuentes efectos adversos.
En la última década, la farmacogenética ha comenzado a dar los primeros pasos en el campo del tratamiento de la EP. En este artículo revisaremos los principales polimorfismos que pueden afectar el efecto de los agentes antiparkinsonianos tratando de integrar el conocimiento de los genes/proteínas implicados con aspectos relativos a la farmacocinética y farmacodinámica de los distintos tratamientos (Tabla 1).
.
| |
Familia D1 |
Familia D2 |
| (estimuladores) |
(inhibidores) |
| Sub-Tipo |
D1 |
D5 |
D2 |
D3 |
D4 |
| D2q |
D2p |
F
U
N
C
I
Ó
N |
- Regula funciones motoras y cardiovasculares
- Participa en la regulación de los Mecanismos Sueño – Vigilia |
- Controla la formación AMPc por estimulación de una o mas isoformas de la adenilciclasa |
- Autoreceptor
- Participa en funciones motoras, en algunos aspectos de la emoción y en la integración y expresión de las conductas motivadas
- Regula la síntesis y la liberación de dopamina, así como la secreción de prolactina |
- Participa en algunos aspectos motores y en conductas asociadas con aspectos motivados
- Involucrado en el trastorno depresivo
- Función moduladora a nivel postsináptico |
- Participa de manera importante en la integración y la expresión de acciones motoras |
- Estimula el Sistema Límbico |
| |
Tabla 1 Subtipos de receptores dopaminérgicos y su función en el sistema nervioso central.
Polimorfismos en los Genes de los Receptores de la Dopamina
La L-Dopa se considera el tratamiento médico más eficaz para la EP. Cuando se administra L-Dopa por vía oral, es absorbida por el intestino, entra en la circulación sanguínea, cruza la barrera hematoencefálica y llega hasta el cerebro, donde es convertida en Dopamina por la L-Aminoácido aromático descarboxilasa (AADC). Como hemos visto, la L-Dopa se administra en combinación con un inhibidor periférico de la descarboxilasa para evitar la conversión de L-Dopa a Dopamina fuera del cerebro (Figura 1). Los efectos beneficiosos de la L-Dopa sobre los síntomas, función motora, actividades de la vida diaria y la calidad de vida son evidentes. De todos modos, existen síntomas que no se controlan satisfactoriamente o no responden en absoluto a la L-Dopa, tales como la inestabilidad postural, y las caídas, los episodios de congelación, la disartria, el estado de ánimo, los trastornos del sueño, la disfunción autonómica y el deterioro cognitivo [9,10]. Además, la toma continuada de L-Dopa durante años se asocia al desarrollo de complicaciones motoras, tales como fluctuaciones y discinesias que pueden conllevar discapacidad severa en algunos pacientes. La frecuencia de estas complicaciones varía entre el 40 y el 50% tras 5 años de tratamiento [9,11]. Además, existe una gran variabilidad interindividual en el tiempo transcurrido desde el inicio de tratamiento con L-Dopa y el inicio de complicaciones motoras, aún tras ajustar el cálculo por dosis acumulada total de L-Dopa. Por tanto, existen factores que influencian la aparición de complicaciones motoras de la L-Dopa que aún no son bien comprendidos. Comprender los mecanismos y factores que influencian la aparición de las complicaciones es esencial para desarrollar estrategias encaminadas a reducir el riesgo de su presentación. Además, el tratamiento con L-Dopa también se asocia a cuadros confusionales, alucinaciones, psicosis, síndrome de disregulación dopaminérgica, punding y trastorno del control de impulsos [9].
Por otro lado, los agonistas dopaminérgicos también estimulan los receptores postsinápticos de Dopamina en el estriado. Se han desarrollado 2 tipos de agonistas dopaminérgicos: los derivados de la ergotamina, como la bromocriptina, pergolida y cabergolina; y los no ergóticos, como el pramipexol, ropinirol, piribedil, apomorfina y rotigotina [9,12]. Estos fármacos pueden usarse en monoterapia o en combinación con L-Dopa o inhibidores de la MAO [9,12,13]. Los efectos secundarios de los agonistas dopaminérgicos incluyen alucinaciones, somnolencia excesiva diaria, ataques del sueño y una variedad de trastornos del control de impulsos, tales como hipersexualidad, juego patológico, compra patológica y alimentación compulsiva y punding [9,12].
Existen 5 receptores de la Dopamina, D1-D5, sobre los que actúa la Levo-Dopa. Sin embargo los agonistas dopaminérgicos actúan sólo sobre algunos de ellos. El gen del receptor D1 se localiza en el cromosoma 5, y se han identificado varios polimorfismos en las regiones 5’ y 3’ del gen, si bien ninguno de estos polimorfismos parece influenciar su unión a sustratos [14]. El gen del receptor D2 se localiza en el cromosoma 11. El polimorfismo TaqIA de un gen próximo a DRD2, denominado ANKK1, podría influenciar la respuesta terapéutica. Los pacientes con el alelo A1 para el polimorfismo eTaqIA tienen menor densidad de receptores D2 en el estriado, y este hecho se asocia a mayor riesgo a presentar fluctuaciones con el tratamiento con L-Dopa [15]. Sin embargo, no se ha encontrado asociación entre los polimorfismos TaqIA y la respuesta terapéutica al agonista dopaminérgico Pramipexol [7]. Sin embargo, otros investigadores no consiguieron replicar el efecto del polimorfismo TaqIA en la variabilidad interindividual en la demanda dopaminérgica en EP [16]. Los portadores del polimorfismo TaqIA son más propensos a desarrollar alucinaciones tardías [17]. Los portadores homocigotos del alelo A1 también se ha visto que son más propensos a padecer ataques súbitos de somnolencia, un fenómeno común en pacientes con EP a tratamiento con agonistas dopaminérgicos [18].
Otro polimorfismo en el gen DRD2, la inserción/delección ins/ del en 141C, en la región promotora DRD2, se ha asociado a mayor densidad estriatal del receptor DRD2 y a incremento en la expresión de mRNA [19]. Por otro lado, la frecuencia del genotipo-141C/C en este gen es mayor en los pacientes con EP que desarrollan alucinaciones como efecto secundario de los tratamientos [17], mientras que los portadores de tandas cortas de repeticiones del dinucleótido intrónico CA presentan con mayor facilidad discinesias derivadas del tratamiento con L-Dopa [20,21].
El polimorfismo Ser9Gly de DRD3 está relacionado con la afinidad de unión de la Dopamina a los receptores D3. Un estudio encontró que los pacientes con genotipo Ser/Ser en el polimorfismo Ser9Gly de DRD3 mostraron mejor respuesta clínica a pramipexol en comparación con los portadores del alelo Gly [7]. Sin embargo otros estudios no alcanzaron las mismas conclusiones [15,22,23], lo que sugiere que estos acontecimientos sean también influenciados por polimorfismos en otros genes.
Polimorfismos en el gen Catechol-Omethyltrasferasa (COMT)
La enzima COMT cataboliza la L-Dopa a 3-O–methyl-dopa. Así, una estrategia terapéutica clásica consiste en inhibir esta enzima de modo que aumente la cantidad de L-Dopa disponible. Desde hace años se han comercializado varios tratamientos para la EP que actúan inhibidores de la COMT en la periferia (entacapone, tolcapone) y en el cerebro (tolcapone) [9,12,24]. La administración de inhibidores de la COMT junto a la L-Dopa, incrementa su vida media y el área bajo la curva plasmática, disminuyendo el tiempo off e incrementando el tiempo ON, lo que resulta de gran utilidad en pacientes con fluctuaciones en la respuesta a L-Dopa [4,12,24]. Los inhibidores de la COMT no están exentos de efectos adversos. Por ejemplo un potencial efecto adverso de la Tolcapona es que puede inducir toxicidad hepática [25].
El gen COMT es diana de la farmacogenética. Se localiza y en cromosoma 22 y codifica para 2 formas distintas de la proteína, una es la forma soluble S-COMT y la otra para la proteína unida a membrana MB-COMT, que es la forma de mayor expresión en el cerebro. Hay 2 alelos del gen COMT que codifican para las variantes termolábil de baja actividad COMT L/L y termoestable de alta actividad H/H, basada en una sustitución de metionina por valina en los codones 108 y 158 de las proteínas S-COMT y MB-COMT respectivamente [26]. Algunos estudios poblacionales concluyen que las frecuencias de estos polimorfismos son similares en la EP y en la población general [27], mientras que otras sí ven diferencias entre enfermos y controles [28]. Además, algunas poblaciones pueden tener diferencias particulares. Por ejemplo en África el alelo COMT L es menos frecuente [29].
En teoría, las diferencias genéticas en la actividad COMT podrían influenciar la respuesta individual a la L-Dopa y el riesgo de desarrollar complicaciones motoras precoces. Así, los pacientes con metabolismo más lento de la L-Dopa podrían ser mejores respondedores al tratamiento, si bien los resultados de los estudios realizados hasta la fecha han sido contradictorios.
La actividad metabólica de COMTL/L probablemente permite un catabolismo enlentecido del fármaco, haciendo que las concentraciones plasmáticas y en el SNC sean más estables. El efecto del alelo COMT L sobre la respuesta clínica a las terapias antiparkinsonianas también ha sido evaluado en un estudio en el que se administraban altas dosis de Piridoxina, un cofactor AADC, encontrándose que la mejor respuesta se producía en los pacientes homocigotos COMTL/L [30]. Algunos grupos no han encontrado diferencias entre los distintos genotipos COMT y la respuesta a la L-Dopa, así como entre los genotipos COMT y la aparición de fluctuaciones wearing-off o discinesias en los estudios de Lee y Watanabe [28,31-33]. Sin embargo, Bialecka et al. [6] han comunicado que los homocigotos COMT L/L podrían beneficiarse de tratamientos más eficientes y seguros con L-Dopa. Los mismos investigadores señalan que los pacientes COMT H/H presentan más frecuentemente discinesias y otras fluctuaciones motoras que aquellos con genotipo COMT L/L [34-36]. Otros estudios han demostrado que el genotipo COMTH/H mejora el efecto de la entacapona sobre la farmacocinética y farmacodinámica de la L-Dopa [37]. Por contra, Chong et al comunicaron que el genotipo de la COMT no es un factor determinante para la respuesta de la tolcapona [38]. Por último, mencionar que los portadores del alelo L (LL y LH) tienen mayor riesgo de presentar somnolencia diurna que los HH [39].
Polimorfismos en los Genes Monoaminooxidasa A y B (MAO-A y MAO-B)
MAO-A y MAO-B producen la oxidación de Dopamina. MAO-A se localiza principalmente en los órganos periféricos donde contribuyen a aproximadamente el 80% de la actividad gastrointestinal y MAO-B es la isoforma principal en el cerebro, especialmente en los ganglios basales. En el cerebro, proceso que puede ser inhibido por los inhibidores de la MAO-B (Selegilina, Rasagilina y Safinamida, éste último inhibidor reversible), incrementando así los niveles de Dopamina disponibles a nivel de sinapsis. Estos tratamientos pueden ser utilizados como monoterapia en fases iniciales de la enfermedad, o más comúnmente en combinación con L-Dopa para reducir las fluctuaciones motoras e incrementar el tiempo on [9,12]. Además, algunos estudios han concluído que estos medicamentos podrían tener cierto efecto neuroprotector [40,41].
Los polimorfismos MAO-A y MAO-B también se han examinado como posibles moduladores de los efectos de los fármacos antiparkisonianos. Los genes de MAO-A y MAO-B están localizados en el cromosoma X [42]. Resulta de especial interés el polimorfismo del intrón 13 (Tsp45I) del gen MAO-B (sustitución A G) que ha sido asociado con mayor actividad de la actividad MAO-B [43], lo que podría afectar a los requerimientos posológicos de los pacientes [6]. Por otro lado se ha visto que el haplotipo AG-HH MAOB-COMT está presente con mayor frecuencia en el subgrupo de pacientes que precisan altas dosis de L-Dopa, mientras que el haplotipo AG-LL MAOB-COMT está presente con mayor frecuencia en el subgrupo de pacientes que precisan dosis bajas de L-Dopa, lo que sugiere que este último haplotipo supone un factor que mejora la eficacia de la L-Dopa en pacientes con EP (Tabla 2) [6].
| |
Eficacia |
Psicosis Alucinaciones |
Discinesias y fluctuaciones motoras |
Somnolencia |
| COMT |
COMT L/L ↑ L-Dopa |
- |
COMT H/H |
COMT L/L y L/H |
| MAO-B |
sustitución A→G Tsp45I ↓ L-Dopa |
- |
- |
- |
| DR |
Ser9Gly-DRD3 ↑ Agonista |
141C/-DRD2
TaqIA-DR2 |
Repet.CA-DRD2 |
- |
Tabla 2 Principales polimorfismos con influencia sobre la eficacia y efectos secundarios de los tratamientos antiparkinsonianos.
Futuro
En primer lugar, es necesario tratar de replicar algunos de los hallazgos reportados hasta la fecha, ya que en ocasiones los resultados no han sido consistentes o incluso han sido contradictorios entre distintos grupos.
Además, hay otros muchos efectos adversos de los fármacos antiparkinsonianos cuyas bases genéticas y asociación a SNP aún no han sido explorados, como el Trastorno de Control de Impulsos (TCI). Es importante abordar el estudio de estos efectos dado que presentan gran repercusión clínica e incluso social.
Igualmente tampoco se ha estudiado hasta la fecha el efecto de los tratamientos dirigidos a controlar los síntomas no motores, como pueden ser los fármacos anticolinesterásicos, para el deterioro cognitivo, o los neurolépticos atípicos para el control de síntomas psicóticos. Tampoco se han estudiado los fármacos antidepresivos y ansiolíticos que con frecuencia se pautan en estos pacientes.
También cabe estudiar la posible relación entre efecto de los fármacos y no ya sobre variables clínicas directas, sino como variables clínicas indirectas, como los biomarcadores. El desarrollo de biomarcadores para la EP y otras enfermedades neurodegenerativas permiten valorar el sustrato neuropatológico de la enfermedad. Por ejemplo, el DaTSCAN permite valorar la integridad de la vía nigroestriada. Ésto nos ofrece una gran oportunidad para valorar el efecto de los fármacos sobre el proceso neurodegenerativo en sí, y poder averiguar si un determinado fármaco tiene, por ejemplo, un efecto neuroprotector.
Además de buscar nuevos estudios de asociación, el futuro vendrá marcado por las nuevas técnicas de genética molecular. A destacar especialmente los estudios de asociación del genoma completo (en inglés, GWAS (Genome-wide association study) o WGAS (Whole genome association study). Se trata de un análisis de una variación genética a lo largo de todo el genoma humano con el objetivo de identificar su asociación a un rasgo observable. Los GWAS suelen centrarse en asociaciones entre los polimorfismos de un solo nucleótido (SNPs) y rasgos como las principales enfermedades. Para ello se deben estudiar una gran cantidad de individuos, de modo que se puedan comparar datos genéticos entre ellos. De este modo, al comparar los datos genéticos obtenidos de las secuenciaciones de los genomas de diferentes individuos, podemos hallar genes ligados a enfermedades o caracteres. Por ejemplo, podemos contrastar cómo quizá se produce la aparición de uno o varios SNPs (variación de un sólo par de bases) o una deleción, repetición, etc. en una secuencia del genoma siempre que aparece el mismo fenotipo, pudiendo así concluir que este cambio a nivel genético se corresponde con un rasgo.
Sorprendentemente, la mayoría de las variaciones de SNPs asociadas con enfermedades no se presentan en las regiones codificantes del ADN, sino que normalmente están en las amplias regiones no codificantes intergénicas o en los intrones que se extraen de la secuencia de ADN cuando las proteínas son procesadas. Estas son presumiblemente secuencias de ADN que controlan otros genes, pero normalmente su función proteica no se conoce.
Por tanto, los estudios farmacogenéticos en la EP no han hecho más que empezar.
Conclusiones
La enfermedad de Parkinson es una enfermedad con una etiología compleja, heterogénea y en parte desconocida. El proceso neurodegenerativo conduce a trastornos del movimiento y otros síntomas no motores. Los tratamientos actualmente existentes, como la Levodopa, agonistas dopaminérgicos, inhibidores de la COMT e inhibidores de la MAO-B van dirigidos a controlar los síntomas motores. Sin embargo, cada paciente puede mostrar distintas respuestas, tanto en términos de eficacia como de efectos secundarios. La farmacogenética nos permite aproximarnos a lass bases moleculares que hay detrás de algunas de estas diferencias terapéuticas al tiempo que establecer estrategias terapéuticas individualizadas que permitan pautas de tratamiento más seguras y eficaces. Son necesarios más estudios para confirmar estas asociaciones y explorar otras asociaciones potenciales, tales como los receptores metabotropicos de glutamato, BDNF y TrkB receptor, NGF, glial cell-line derived neurotrophic factors and FGF-2.
11133
References
- Lazarou J, Pomeranz B, Core P (1998) Incidence of adverse drug reaction in hospitalized patients. A metaanalisis of prospective studies JAMA279: 1200-1205.
- Vilà A, San José A, Roure C, Armadans L, Vilardell M (2003) Grupo para el estudio delas Reacciones Adversas a Medicamentos en pacientes mayores hospitalizados.Med Clin (Barc) 120:613-618.
- O’Shaughnessy KM (2006) HapMap, pharmacogenomics,and the goal of personalized prescribing. Br J Clin Pharmacol61:783-786.
- de Lau LM, Breteler MM (2006) Epidemiology of Parkinson's disease. Lancet Neurol5:525-535.
- Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18:R48-R59.
- Białecka M, Droździk M, Kłodowska-Duda G, Honczarenko K, Gawrońska-Szklarz B, et al. (2004) The effect of monoamine oxidase B (MAOB) and catechol-O-methyltransferase (COMT) polymorphisms on levodopa therapy in patients with sporadic Parkinson’s disease. Acta Neurol Scand 110: 260-266.
- Liu YZ, Tang BS, Yan XX, Liu J, Ouyang DS, et al. (2009) Association of the DRD2 and DRD3 polymorphisms with response to pramipexole in Parkinson’s disease patients. Eur J Clin Pharmacol65:679-683.
- Benbir G, Ózemecki S, Sinar M, Beskardes F, Apaydin H, et al. (2006) Features associated with the development of hallucinations in Parkinson's Disease. Acta Neurologica Scandinavica 114:239-243.
- Olanow C, Stern M, Sethi K (2009) The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72:S1-S136.
- Sethi K (2008) l-Dopa unresponsive symptoms in Parkinson disease. Mov Disord 23: S521-S533.
- Fox S, Lang A (2008) l-Dopa-related motor complications-phenomenology. Mov Disord 23: S509-S514.
- Factor S (2008) Current status of symptomatic medical therapy in Parkinson’s disease. Neurotherapeutics 5: 164-180.
- Bonuccelli U, Del Dotto P, Rascol O (2009) Role of dopamine receptor agonists in the treatment of early Parkinson’s disease. Parkinsonism Relat Disord 15: S44-S53.
- Arbouw ME, van Vugt JP, Egberts TC, Guchelaar HJ (2007) Pharmacogenetics of antiparkinsonian drug treatment: a systematic review.Pharmacogenomics 8: 159-176.
- Wang J, Liu ZL, Chen B (2001) Association study of dopamine D2, D3 receptor gene polymorphisms with motor fluctuations in PD.Neurology 56: 1757-1759.
- Paus S, Grünewald A, Klein C, Knapp M, Zimprich A, et al. (2008) The DRD2 TaqIA polymorphism and demand of dopaminergic medication in Parkinson’s disease. Mov Disord 23: 599-602.
- Makoff AJ, Graham JM, Arranz MJ, Forsyth J, Li T, et al. (2000)Association study of dopamine receptor gene polymorphisms with drug-induced hallucinations in patients with idiopathic Parkinson's disease. Pharmacogenetics 10: 43-48.
- Rissling I, Geller F, Bandmann O, Stiasny-Kolster K, Körner Y, et al. (2004) Dopamine receptor gene polymorphisms in Parkinson’s disease patients reporting “sleep attacks”. Mov Disord 19: 1279-1284.
- Jonsson EG, Nothen MM, Grunhage F, Farde L, Nakashima Y, et al. (1999) Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry 4: 290-296.
- Zappia M, Annesi G, Nicoletti G, Arabia G, Annesi F, et al. (2005) Sex differences in clinical and genetic determinants of l-Dopa peak-dose dyskinesias in Parkinson disease; an exploratory study. Arch Neurol 62: 601-605.
- Strong JA, Dalvi A, Revilla FJ, Sahay A, Samaha FJ, et al. (2006) Genotype and smoking history affect risk of l-Dopa-induced dyskinesias in Parkinson’s disease.Mov Disord 21: 654-659.
- Kaiser R, Hofer A, Grapengiesser A, Gasser T, Kupsch A, et al. (2003) l-dopa-induced adverse effects in PD and dopamine transporter gene polymorphism.Neurology 60: 1750-1755.
- Paus S, Gadow F, Knapp M, Klein C, Klockgether T, et al. (2009) Motor complications in patients form the German Competence Network on Parkinson’s disease and the DRD3 Ser9Gly polymorphism. Mov Disord24: 1080-1084.
- Bonifácio MJ, Palma PN, Almeida L, Soares-da-Silva P (2007) Catechol-O-methyltransferase and its inhibitors in Parkinson’s disease. CNS Drug Rev 13: 352-379.
- Assal F, Spahr L, Hadengue A, Rubbia-Brandt L, Burkhard PR (1998) Tolcapone and fulminant hepatitis. Lancet 352: 958.
- Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melén K, et al. (1995) Kinetics of human soluble and membrane-bound catechol-O-methyltransferase;a revised mechanism and description of the termolabile variant of the enzyme. Biochemistry 34: 4202-4210.
- Syvänen C, Tilgmann C, Rinne J, Ulmanen I (1997) Genetic polymorphism of catechol-O-methyltransferase (COMT): correlation of genotype with individual variation of S-COMT activity and comparison of the allele frequencies in the normal population and Parkinsonian patients in Finland. Pharmacogenetics 7: 65-71.
- Watanabe M, Harada S, Nakamura T, Ohkoshi N, Yoshizawa K, et al. (2003) Association between catechol-O-methyltransferase gene polymorphisms and wearing-off and dyskinesia in Parkinson’s disease. Neuropsychobiology 48: 190-193.
- Espinoza S, Manago F, Leo D, Sotnikova TD, Gainetdinov RR (2012) Role ofcatechol-O-methyltransferase (COMT)-dependent processes in Parkinson's diseaseand L-DOPA treatment. CNS Neurol Disord Drug Targets11:251-263.
- Tan EK, Cheah SY, Fook-Chong S, Yew K, Chandran VR, et al. (2005) Functional COMT variant predicts response to high dose pyridoxine in Parkinson’s disease.Am J Med Genet B Neuropsychiatr Genet 137: 1-4.
- Lee MS, Lyoo ChH, Ulmanen I, Syvanen AC, Rinne JO (2001) Genotypes of catechol-O-methyltransferase and response to l-Dopa treatment in patients with Parkinson’s disease. Neurosci Lett 298: 131-134.
- Contin M, Martinelli P, Mochi M, Riva R, Albani F, et al. (2005) Genetic polymorphism of catechol-O-methyltransferase and levodopa pharmacokinetic-pharmacodynamic pattern in patients with Parkinson’s disease. Mov Disord 20: 734-739.
- Yin B, Chen Y, Zhang L (2013) Association Between Catechol-O-Methyltransferase(COMT) Gene Polymorphisms, Parkinson's Disease, and Levodopa Efficacy. Mol Diagn Ther 18: 253-260.
- Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Tan EK, et al. (2008) The association of functional catechol-O-methyltransferase haplotypes with risk of Parkinson’s disease, levodopa treatment response, and complications. Pharmacogenet Genomics 18: 815-821.
- Rivera-Calimlim L, Reilly DK (1984) Difference in erythrocyte catechol-O-methyltransferase activity between Orientals and Caucasian; difference in l-Dopa tolerance. Clin Pharmacol Ther 35: 804-849.
- Routtinen HM (1998) COMT genotypes and response to l-Dopa in Parkinson’s disease; influence of COMT inhibition with entacapone. Mov Disord 13: 120.
- Corvol JC, Bonnet C, Charbonnier-Beaupel F, Bonnet AM, Fiévet MH, et al. (2011) The COMT Val158Met polymorphism affects the response to entacapone in Parkinson’s disease: a randomized crossover clinical trial. Ann Neurol 69: 111-118.
- Chong DJ, Suchowersky O, Szumlanski C, Weinshilboum RM, Brant R, et al. (2000) The relationship between COMT genotype and the clinical effectiveness of tolcapone, a COMT inhibitor, in patients with Parkinson’s disease. Clin Neuropharmacol23:143-148.
- Frauscher B, Hogl B, Maret S,Wolf E, Brandauer E, et al. (2004) Association of daytime sleepiness with COMT polymorphism in patients with parkinson disease; a pilot study. Sleep 27: 733-736.
- Hauser RA, Lew MF, Hurtig HI, Ondo WG, Wojcieszek J, et al. (2009) Long-term outcome of early versus delayed rasagiline treatment in early Parkinson’s disease.Mov Disord 24: 564-573.
- Olanow CW, Rascol O, Hauser R, Jankovic J, Lang A, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med361: 1268-1278.
- Hsu YP, Powell JF, Sims KB, Breakefield XO (1989) Molecular genetics of monoamine oxidases. J Neurochem 53: 12-18.
- Balciuniene J, Emilsson L, Oreland L, Pettersson U, Jazin E (2002) Investigation of the functional effect of monoamine oxidase polymorphisms in human brain. Hum Genet 110: 1-7.

