Key words
|
| |
| Lornoxicam, Sustained release drug delivery system, Matrix tablets, HPMC |
| |
Introduction
|
| |
| The term arthritis means "Joint inflammation" but is generally used to describe inflammatory and degenerative conditions of the joints. Contrary to popular misconception, arthritis is not a disease, which is inevitable with old age, it can affect at any age. Also, there are hundred different kinds of arthritis, the most common of which is the osteoarthritis, rheumatoid arthritis and gout.(1) Lornoxicam is a non-steroidal anti-inflammatory drug with analgesic property and belongs to the class Oxicams. Lornoxicam inhibits synthesis of prostaglandins via inhibition of cyclo-oxygenase enzyme, but does not inhibit 5-lipo oxygenase. In vitro the inhibition of cyclo oxygenase does not result in an increase in leukotrienes formation.(39) |
| |
MATERIALS AND METHODS
|
| |
 |
| |
FORMULATION OF SUSTAINED RELEASE
|
| |
| MATRIX TABLETS OF LORNOXICAM: |
| |
| Matrix tablets of Lornoxicam with other excipients were prepared by direct compression. The weight of Lornoxicam was kept constant in all the prepared tablets at 8 mg/tablet. Different viscosity grades of HPMC namely HPMC K4M, HPMC K15M, HPMC K100M were chosen as polymeric matrix materials. Micro crystalline cellulose (MCC) was selected as tablet diluent for increasing the compressibility and flowability of the ingredients as well as to maintain the tablets at constant weight of 120 mg. Magnesium stearate was used as a lubricant at concentration of 2% by weight of tablet. To make powder mixtures, the drug, polymer and MCC were thoroughly mixed for 30 min by means of pestle and mortar. This powder mixture was then lubricated with magnesium stearate then compressed into tablets in 6 mm rotary tablet punching machine. The force of compression was adjusted so that hardness of all the prepared tablets ranges from 5.5-6.5 kg/cm . The detailed compositions of the prepared matrix tablets formulations are given in (Table 4). |
| |
EVALUATION OF PREFORMULATION PARAMETERS :-
|
| |
|
i) Micromeritic properties44:-
|
| |
|
a) Angle of repose:-
|
| |
| The angle of repose of powder was determined by the funnel method. The accurately weighed powder was taken in a funnel. The height (h) of the funnel was adjusted in such a way that the tip of the funnel just touches the apex of the heap of the powder. The powder was allowed to flow through funnel freely onto the surface. The diameter of the powder cone was measured and angle of repose was calculated using the following equation. |
| |
| tan θ = h/r (1) |
| |
| Therefore,θ = tan h/r |
| |
| Where, θ = angle of repose, |
| |
| h = height of the pile, |
| |
| r = radius of the pile base |
| |
|
b) Bulk density :-
|
| |
| Both loose bulk density (LBD) and tapped bulk density (TBD) were determined. Powder from each formulation, previously lightly shaken to break any agglomerates formed was introduced into a 10 ml measuring cylinder. After the initial volume was observed, the cylinder was allowed to fall under its own weight onto a hard surface from the height of 2.5 cm at 2 sec intervals. The tapping was continued until no further change in volume was noted. Bulk density is calculated by using formula: |
| |
| Weight of the powder Bulk density (pb) = Bulk volume of the powder |
| |
| Weight of the powder Tapped density (p,) = Tapped volume of the powder |
| |
|
c) Carr’s index:-
|
| |
| It helps in measuring the force required to break the friction between the particles and the hopper. It is expressed in % and given by:- |
| |
| Carr’s index (%) = [(TBD - LBD) x 100]/TBD Where, LBD = weight of the powder/volume of the packing TBD = weight of the powder/tapped volume of the packing |
| |
|
ii) Physicochemical parameters:-
|
| |
|
a) Tablet hardness44 :-
|
| |
| |
| The resistance of tablet for shipping or breakage, under conditions of storage, transportation and handling, before usage, depends on its hardness. The hardness of tablet of each formulation was measured by using Pfizer hardness tester. |
| |
| Thickness of tablets was important for uniformity of tablet size. Thickness was measured by using screw gauze on 3 randomly selected samples. |
| |
|
c) Friability44 :-
|
| |
| Friability is the measure of tablet strength. Roche Friabilator was used for testing the friability using the following procedure. Twenty tablets were weighed accurately and placed in the plastic chamber that revolves at 25 rpm for 4 mins dropping the tablets through a distance of six inches with each revolution. After 100 revolutions the tablets were reweighed and the percentage loss in tablet weight was determined. |
| |
| % loss =(Initial wt. of tablets - Final wt. of tablets)/Initial wt. of tablets*100 |
| |
|
d) Weight variation45:-
|
| |
| Twenty tablets were weighed individually and the average weight was determined. Then percentage deviation from the average weight was calculated. According to IP standards, not more than two of the individual weight deviates from the average weight by more than the percentage shown in the (Table 7) and none deviates by more than twice that percentage. |
| |
|
e) Uniformity of drug content45 :-
|
| |
| Ten tablets were weighed and average weight is calculated. All tablets were crushed and powder equivalent to 8 mg drug was dissolved in 8 ml of 0.1N NaOH and the volume was made upto 100 ml with pH 6.8 phosphate buffer. The solution was shaken for 1 h and kept for 24 h. From the stock solution, 1 ml solution was taken in 10 ml volumetric flask and the volume was made with pH 6.8 phosphate buffer. Solution was filtered and absorbance was measured spectrophotometrically at 379 nm against pH 6.8 phosphate buffer as a blank. Amount of drug present in one tablet was calculated. |
| |
|
f) Dissolution studies21, 46:-
|
| |
| The release rate of Lornoxicam from sustained matrix tablets were determined using USP dissolution testing apparatus II (paddle type) at 50 rpm. The dissolution test was performed using 750 ml of 0.1 N HCl (pH 1.2) for 2 h at 37 ± 0.5 °C and then 250 ml of 0.2 M tri sodium phosphate (Na3PO4.12H2O) was added and pH is adjusted to 6.8 as described in the USP 26/NF monograph. Dissolution test was carried out for a period of 12 h using 0.1N HCl (pH 1.2) for first 2 h and then the pH is adjusted to 6.8 for the rest of the period. The temperature of the dissolution medium is maintained at 37±0.5°C. 10 ml of the sample was withdrawn at regular intervals and replaced with the same volume pre-warmed with fresh dissolution medium. After filtration, the amount of drug release was determined from the standard calibration curve of pure drug. |
| |
|
KINETICS MODELLING OF DRUG DISSOLUTION PROFILE7:
|
| |
| The dissolution profile of most satisfactory formulation was fitted to zero order, first order and Higuchi model to ascertain the kinetic modeling of the drug release. The methods were adopted for deciding the most appropriate model |
| |
| 1. Cumulative percent drug released versus time (Zero order kinetic model) |
| |
| 2. Log cumulative percent drug remaining versus time (First order kinetic model) |
| |
| 3. Cumulative percent drug released versus square root of time (Higuchi’s model). |
| |
|
> Zero order:
|
| |
| In many of the modified release dosage forms, particularly sustained or controlled release dosage forms (those dosage forms that release the drug in planned, predictable and slower than the normal manner) is zero order kinetic. The plot of cumulative percent drug released versus time is the linear. |
| |
|
> First order:
|
| |
| Most conventional dosage forms exhibits this dissolution mechanism. Some modified release preparation, particularly prolonged release formulations, adheres to this type of dissolution pattern. It assumes that the drug molecules, diffuses out through a gel like layer formed around the drug during the dissolution process. A plot of log cumulative percent drug remaining versus time is the linear. |
| |
|
> Higuchi model:
|
| |
| A large number of modified release dosage form contain some sort of matrix system. In such instances, the drug dissolves from the matrix. The dissolution pattern of the drug is dictated by water penetration rate (diffusion controlled). In Higuchi model, a plot of cumulative percent drug released versus square root of time is linear. |
| |
4) STABILITY STUDIES FOR THE MOST SATISFACTORY FORMULATION OF SUSTAINED RELEASE MATRIX TABLETS OF LORNOXICAM47:
|
| |
| Stability testing of drug products begins as a part of drug discovery and ends with the demise of the compound or commercial product. To assess the drug and formulation stability, stability studies were done according to ICH guidelines. The stability studies were carried out of the most satisfactory formulation as per ICH guidelines. The most satisfactory formulation sealed in aluminum packaging and kept in humidity chamber maintained at 30 ± 2 °C / 65 ± 5 % RH and 40 ± 2 °C / 75 ± 5 % RH for two months. At the end of studies, samples were analyzed for the drug content, in vitro dissolution, sustained behavior and other physicochemical parameters. |
| |
RESULTS
|
| |
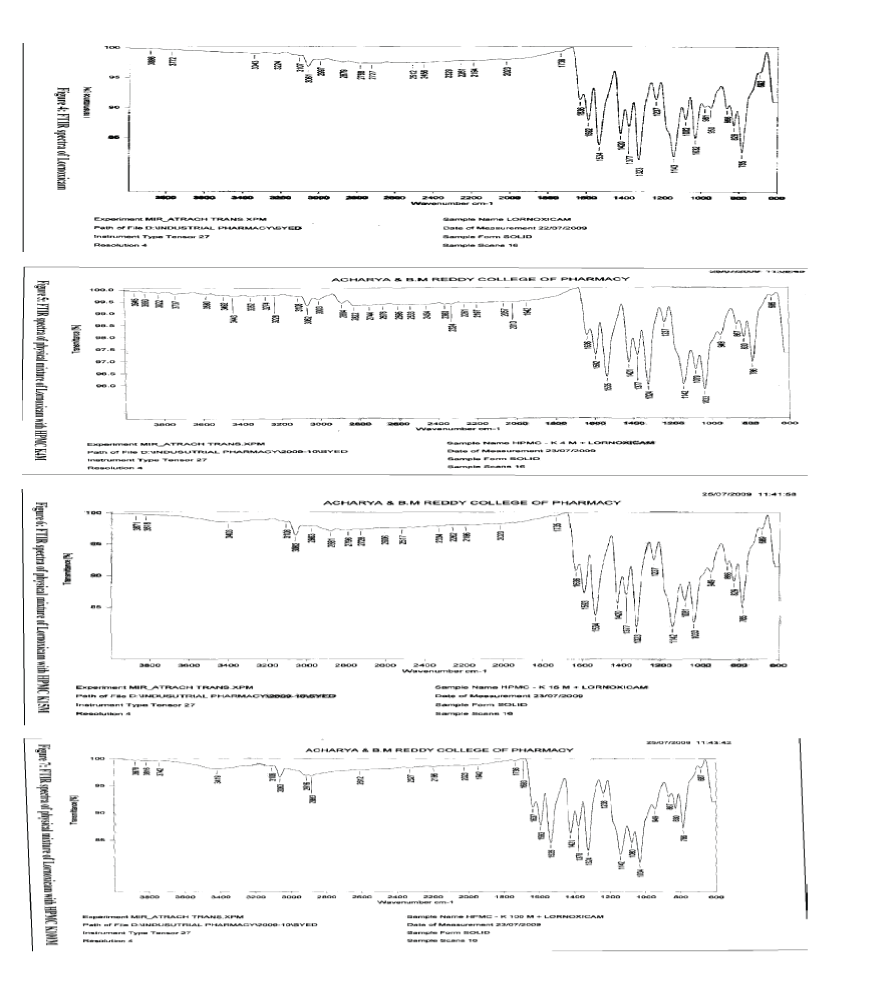
FTIR OF PURE LORNOXICAM DRUG48:
|
| |
 |
| |
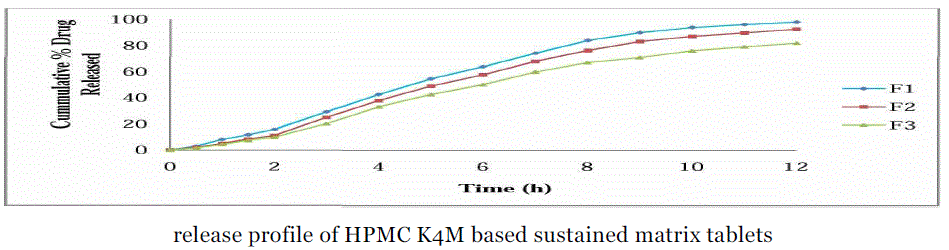
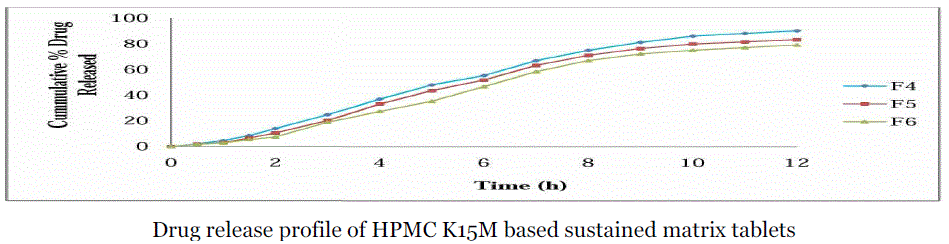
| Comparative drug release profile of Lornoxicam matrix tablets:- |
| |
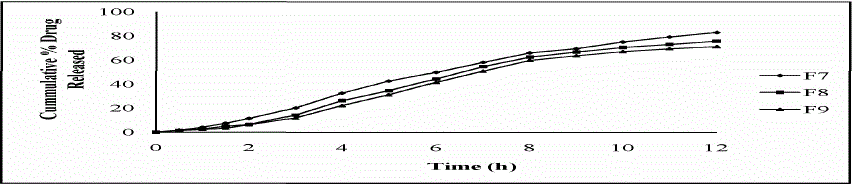
| 1) Comparative dissolution studies of formulation F1, F2 & F3:- |
| |
 |
| |
| 2) Comparative dissolution studies of formulation F4, F5 & F6:- |
| |
 |
| |
 |
| |
 |
| |
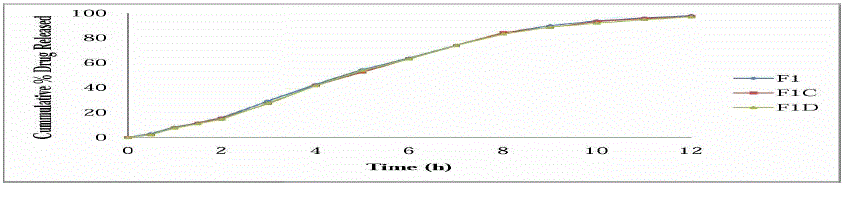
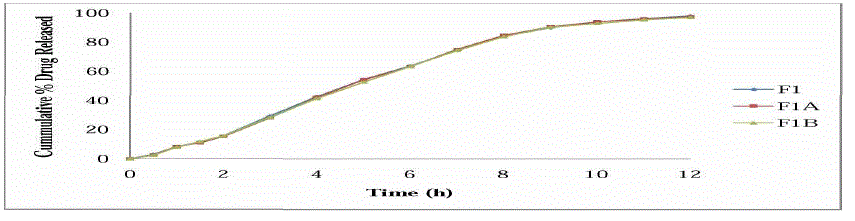
STABILITY STUDIES :-
|
| |
| All values are mean of 3 readings |
| |
| A, C = 30±2°C/65±5%RH |
| |
| B, D = 40±2°C/75±5%RH |
| |
 |
| |
 |
| |
DISCUSSION
|
| |
| In the present study, Lornoxicam matrix tablets were prepared by using HPMC (K4M, K15M, K100M) as a drug retardant polymer. A total number of nine formulations were prepared by direct compression technique. The pre formulation studies such as bulk density, tapped density, angle of repose and carr s index evaluated were found to be within prescribed limits and indicated good free flowing property. The data obtained from physicochemical parameters such as hardness, friability, weight variation, drug content and in vitro drug dissolution are shown in (Table 11, 12, 13). |
| |
PREFORMULATION STUDIES:-
|
| |
| Estimation of Lornoxicam was carried out by SHIMADZU-1700 UV spectrophotometer at Amax 373 nm & 379 nm in simulated gastric fluid 0.1N HCl (pH 1.2) and simulated intestinal fluid (pH 6.8). The linear coefficients of each were found closer to 1. By using the regression coefficient equation the assay and % CDR were calculated. |
| |
|
UV spectrum analysis of Lornoxicam:
|
| |
| At the outset, method for the estimation for the drug was developed. Lornoxicam showed maximum absorption at wavelength 373 nm in O.IN HCl (pH 1.2) and 379 nm in phosphate buffer (pH 6.8). Standard calibration curve obeyed Beer’s law at given concentration range of 3 ug/ml to 18 (ig/ml and when subjected to regression analysis, the value of regression coefficient was found to be 0.998, which showed linear relationship between concentration and absorbance. |
| |
| Any formulation development work has to be preceeded by preformulation studies. This preformulation study includes drug polymer compatibility study and analytical investigation of drug. FTIR study showed that there is no interaction between drug and polymer. So, the drug and polymer are compatible. |
| |
|
FORMULATION STUDIES
|
| |
| Various formulations of sustained release matrix tablets were developed for Lornoxicam by using selected polymers like HPMC K4M, HPMC K15M, HPMC K100M. Microcrystalline cellulose was used as filler and magnesium stearate was used as lubricants. Various formulations of sustained release matrix tablets were prepared by direct compression technique using 6 mm flat punches to an average weight of 120 mg. |
| |
MICROMERITIC PROPERTIES:
|
| |
|
Angle of repose:
|
| |
| The results of angle of repose were ranged between 24.78° ± 1.15 to 29.58° ± 0.44 (Table 11) which indicates good flow properties of powder. |
| |
|
Compressibility index:
|
| |
| The compressibility index values were found to be in the range of 15.08 ± 1.38% to 19.84 ± 2.75% (Table 11). These findings indicated that the powder mixture of all batches of formulation exhibited good flow characters and hence, were suitable for direct compression into matrix tablets. |
| |
EVALUATION OF PHYSICOCHEMICAL PARAMETERS: -Tablet Hardness:
|
| |
| Hardness of the developed formulations F1 to F9 varied from 6.0 ± 0.11 to 6.6 ± 0.46 kg/cm2 (Table 12) in all the formulation indicating good mechanical strength with an ability to withstand physical and mechanical stress condition while handling. |
| |
|
Tablet Thickness:
|
| |
| Thickness of the developed formulations F1 to F9 varied from 3.04 ± 0.032 mm to 3.53 ± 0.174 mm (Table 12) in all the formulation and the average thickness is within the range of ± 5%. Each sample was analyzed in triplicate. |
| |
|
Friability:
|
| |
| The loss in total weight of the tablets due to friability was in the range of 0.34 ± 0.05% to 0.78 ± 0.13% (Table 12) in all the formulation and the friability value is less than 1% which ensures that formulated tablets were mechanically stable. |
| |
|
Weight variation:
|
| |
| The maximum % deviation was found to be ± 1.55% (Table 12) from all the formulations. As none of the formulation showed a deviation of more than ± 7.5% (I.P. limit) for any of the tablets tested, the prepared formulations comply with the weight variation test, thus it fulfills the I.P. requirements. |
| |
|
Uniformity of drug content:
|
| |
| The drug content in different tablet formulations was highly uniform and in the range of 94.24 ± 0.89% to 99.31 ± 0.68% (Table 12). The maximum % drug content for all the formulation was found to be 99.31 ± 0.68%. The minimum % drug content for all the formulation was found to be 94.24 ± 0.89%. It is in the limits specified by IP (i.e. ±. |
| |
|
IN VITRO DRUG DISSOLUTION STUDY:
|
| |
| The release of Lornoxicam from sustained release matrix tablets varied according to the types and proportion of matrix forming polymers. |
| |
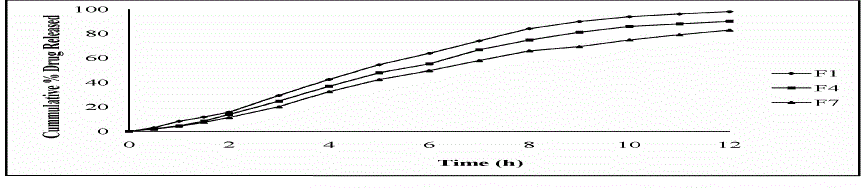
| Ideally, a sustained release tablet should release the required quantity of drug in order to maintain an effective drug plasma concentration. From in vitro drug dissolution profile of Lornoxicam matrix tablet, it was found that 15.81 ± 0.73% of the drug was released till 2 h from F1 formulation (Drug: HPMC 1:1). During 2 to 8 h the marked percentage release was found to be 10-12%. After 8 h more than 60-80% of the drug was released. After 8 h the release rate decreased slightly and a sustained release pattern was observed for 12 h. The hydrophilic matrix of HPMC controlled the Lornoxicam release effectively for 12 h. It was observed that formulation with the drug polymer ratio 1:1 (F1, F4, F7) showed high drug release rates in the range of 98.19 ± 0.49% to 83.53 ± 0.73% when compared to 1:2 ratio (F2, F5, F8) which showed a drug release rates from 93.53 ± 1.21% to 76.64 ± 1.21% and those of 1:3 ratio (F3, F6, F9) which showed a drug release rates in the range of 83.02 ± 1.47% to 71.83 ± 0.73% over a period of 12 h. The order of drug release from the selected polymers were found to decrease in the following order.HPMC K4M > HPMC K15M > HPMC K100M Among the three grades of polymer used the tablets prepared with lower viscosity grade i.e. HPMC K4M, have shown drug release rate (98.19 ± 0.49% to 83.02 ± 1.47% ) and the higher viscosity grade polymers i.e. HPMC K15M (90.78 ± 0.71% to 80.10 ± 1.21%) and HPMC K100M (83.53 ± 0.73% to 71.83 ± 0.73%). But the much difference was not found in the drug release profiles of tablets prepared with HPMC K4M and HPMCK15M |
| |
|
KINETICS MODELING OF DRUG DISSOLUTION PROFILES:
|
| |
| The in vitro release data obtained were fitted into various kinetic models. Correlation coefficients of formulation F1 batch showed higher correlation with zero order plots than higuchi and first order. So, predominant drug release mechanism is controlled release. |
| |
STABILITY STUDIES :-
|
| |
| Stability studies were carried out of the most satisfactory formulation F1, at 30 ± 2°C / 65 ± 5% RH and 40 ± 2°C / 75 ± 5% RH for two months to assess their long term stability as per ICH guidelines. At various time intervals of 30 days and 60 days, samples were evaluated. There was no major change in the various physicochemical parameters evaluated like hardness, drug content, in vitro dissolution pattern at the various sampling points. There was no statistically significant difference between the initial values and the results obtained during stability studies. |
| |
CONCLUSION
|
| |
| Lornoxicam is a non-steroidal anti-inflammatory drug with analgesic property which is used for the better treatment of arthritis. Moreover, the site of absorption of Lornoxicam is in the intestine and has a short half life of 3 to 4 h. Therefore, the present investigation was concerned with the development of the sustained release matrix tablets, which after oral administration were designed to prolong the duration of action. Various formulations were developed by using release rate controlling and gel forming polymers like HPMC (K4M, K15M, K100M) in single by direct compression method. Different proportion of HPMC was associated with decrease in the overall cumulative drug release rate. The higher viscosity polymer had been seen to inhibit the initial burst release of Lornoxicam. Thus, we conclude that from among all the developed formulations, F1 formulation sustained the drug release for longer period of time over 12 h when compare to other formulations. So, F1 was selected as the best formulation. From the result, it is evident that HPMC by forming a matrix retards the release rate of drug and the tablet made by using HPMC can be used as sustained release dosage form. Thus, the objective of the present work was formulating a sustained release dosage form of Lornoxicam by using different proportions and grades of release rate controlling and gel forming polymers like HPMC has been achieved with success. The method of direct compression utilizes minimum machinery and man power. From the economical point of view, it may be beneficial for the local pharmaceutical firms to adopt such simple technologies for the preparation of sustained release product. |
| |
Conflict of Interest
|
| |
| NIL |
| |
Source of Support
|
| |
| NONE |
| |
Tables at a glance
|
 |
|
|
|
| Table 1 |
Table 2 |
Table 3 |
Table 4 |
|
| |
|
|
|
|
| Table 5 |
Table 6 |
Table 7 |
Table 9 |
|
| |


