Keywords
Nifedipine; Eudragit; HPMC; Suatained release; Martix tablet
Introduction
Sustained release dosage form is a modified dosage form that prolongs the therapeutic activity of the drug. Sustained release products provide an immediate release of drug that promptly produces the desired therapeutic effect which is followed by gradual release of additional amounts of drug to maintain this effect over a predetermined period of time. sustained release products often times eliminates the need for night dosing, which benefits not only the patients but the care given as well because of the sustained plasma drug levels [1,2].
The basic rationale of a sustained drug delivery system is to optimize the Pharmacodynamics and Pharmacokinetic properties of a drug in such a way that its utility is maximized through reduction in side effects and cure or control of condition in the shortest possible time by using smallest amount of drug which is administered by the most effective route. Oral route has been the most popular and widely used for sustained delivery of drugs because of convenience and ease of administration, greater flexibility in dosage form design, ease of production and low cost.
Most conventional oral drug products, such as tablets and capsules, are formulated to release the active drug immediately after oral administration, to obtain fast and complete systemic drug absorption. Such immediaterelease products results in rapid drug absorption and onset of action. Plasma drug concentration reduces according to the drug's pharmacokinetic profile after absorption of the drug from the dosage form is absolutely complete. Plasma drug concentrations fall below the minimum effective plasma concentration (MEC), resulting in failure of therapeutic efficacy. Before this point is attained, the other dose is usually given if a sustained therapeutic outcome is desired. The other way of administering dose is to utilize a dosage form that will provide sustained release of drug by maintaining the plasma drug concentrations [3,4].
Materials and Methods
Nifedipine, HPMC-E5, HPMCK100, Eudragit, Magnesium Stearate Lactose, Micro crystalline Cellulose
Wet granulation technique
Steps Involved In Wet Granulation Method for Preparation of Tablets
1. Mixing of the drug(s) and excipients
2. Preparation of binder solution
3. Mixing of binder solution with powder mixture to form wet mass
4. Coarse screening of wet mass using a suitable sieve (6-12 screens)
5. Drying of moist granules
6. Screening of dry granules through a suitable sieve (14-20 screen)
7. Mixing of screened granules with glidant and lubricant (Table 1).
| INGREDIENTS |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
| NIFEDIPINE(mg) |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
| HPMCE5(mg) |
10 |
20 |
30 |
- |
- |
- |
- |
- |
- |
| HPMCK100(mg) |
- |
- |
- |
10 |
20 |
30 |
- |
- |
- |
| EUDRAGIT(mg) |
- |
- |
- |
- |
- |
- |
10 |
20 |
30 |
| Mg.STEARATE(mg) |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
| LACTOSE(mg) |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
| MCC(mg) |
108 |
98 |
88 |
108 |
98 |
88 |
108 |
98 |
88 |
Table 1: Nifedipine formulation.
Results
API characterization
The flow properties of the prepared blend were studied with given IP limits. The hausner’s ratio [5] was found to be excellent and passable in the range of 1.09-1.33 and compressibility index in range of 8.6%-25% (Tables 2 and 3).
| s.no |
API Characterisation |
results |
| 1 |
Physical Appearance |
Nifedipine is a yellow crystalline substance |
| 2 |
Melting point |
172-174°°C |
| 3 |
Solubility |
It is freely soluble in ethanol |
Table 2: API characterisation.
| FORMULA |
ANGLEOF REPOSE |
BULK DENSITY (gm/ml) |
TAPPED DENSITY (gm/ml) |
CARR’S INDEX % |
HAUSNER’S RATIO |
| F1 |
26.6 ±0.2 |
0.555 ±0.1 |
0.714 ±0.1 |
22.22 |
1.285 |
| F2 |
26.0 ±0.3 |
0.384 ±0.4 |
0.434 ±0.3 |
11.53 |
1.130 |
| F3 |
26.7 ±0.4 |
0.416 ±0.2 |
0.476 ±0.3 |
12.50 |
1.142 |
| F4 |
25.1 ±0.1 |
0.476 ±0.3 |
0.526 ±0.2 |
9.52 |
1.105 |
| F5 |
28.3 ±0.4 |
0.625 ±0.1 |
0.833 ±0.1 |
25.00 |
1.333 |
| F6 |
24.4 ±0.4 |
0.521 ±0.3 |
0.631 ±0.3 |
17.39 |
1.121 |
| F7 |
27.1 ±0.1 |
0.588 ±0.3 |
0.666 ±0.4 |
11.76 |
1.333 |
| F8 |
25.2 ±0.1 |
0.277 ±0.2 |
0.312 ±0.2 |
11.11 |
1.133 |
| F9 |
23.2 ±0.2 |
0.434 ±0.2 |
0.476 ±0.3 |
8.695 |
1.095 |
Table 3: Pre compression parameters.
Post compression parameters
The hardness was constantly maintained between 4-6 kg/cm2 for all formulations during compression. Shows the friability values all the formulations [6]. The results indicated that the % friability was between the ranges. The low values of friability indicate that tablets were mechanically hard enough. The drug content of tablets ranged between 97 and 100. The weight variation of all formulations was in the range of 197.6 ± 1.15 to 203.8 ± 1.12 (Table 4).
| FORMULA |
THICKNESS (mm) |
HARDNESS (Kg/cm2) |
%FRIABILITY (w/w) |
WEIGHT VARIATION (mg) |
DRUG CONTENT (%) |
| F1 |
3.2 ± 0.05 |
4.0 ± 0.1 |
0.24 ± 0.03 |
201.8 ± 1.02 |
99.47 ± 0.3 |
| F2 |
3.1 ± 0.02 |
5.5 ± 0.1 |
0.07 ± 0.02 |
203.8 ± 1.12 |
98.89 ± 0.5 |
| F3 |
3.3 ± 0.07 |
5.0 ± 0.4 |
0.19 ± 0.03 |
202.4 ± 1.02 |
98.35 ± 0.9 |
| F4 |
3.2 ± 0.03 |
4.5 ± 0.2 |
0.07 ± 0.01 |
202.1 ± 1.15 |
97.12 ± 0.5 |
| F5 |
3.2 ± 0.04 |
5.3 ± 0.1 |
0.16 ± 0.04 |
199.1 ± 1.13 |
97.56 ± 0.6 |
| F6 |
3.1 ± 0.02 |
5.4 ± 0.3 |
0.07 ± 0.03 |
198.1 ± 1.15 |
98.35 ± 0.3 |
| F7 |
3.3 ± 0.02 |
5.5 ± 0.4 |
0.07 ± 0.01 |
198.6 ± 1.15 |
98.40 ± 0.4 |
| F8 |
3.2 ± 0.05 |
5.4 ± 0.2 |
0.24 ± 0.02 |
197.6 ± 1.15 |
97.84 ± 0.3 |
| F9 |
3.2 ± 0.03 |
5.7 ± 0.2 |
0.04 ± 0.01 |
201.6 ± 1.15 |
99.64 ± 0.1 |
Table 4: Post compression parameters.
In vitro dissolution studies
The dissolution conditions used for studying the drug release from Dosage form:
Apparatus: USP apparatus II (Paddle),
Agitation speed (rpm): 100 rpm,
Medium: 6.8 pH phosphate buffer
Volume: 900 ml
Temperature: 37.0 ± 0.5°C
Time: 1, 4, 8, 12, 16 and 20 hrs.
Wavelength: 238 nm
The samples were withdrawn at predetermined time points, and were analyzed spectrophotometrically at 238 nm(Figure 1). The prepared tablets were subjected to dissolution studies in order to know the amount of drug released and the results of percentage drug release are shown in Table 5. As the concentration of polymer increased, the drug release decreased [7]. In vitro drug release studies revealed that release of Nifedipine from different formulations varies with characteristics and composition of matrix forming polymers. The release rate increased with decreasing concentration of HPMC. These findings are in compliance with the ability of HPMC to form complex matrix network which leads to delay in release of drug from device. Eudragit is hydrophobic polymer which delays the drug release in F7 to F9.
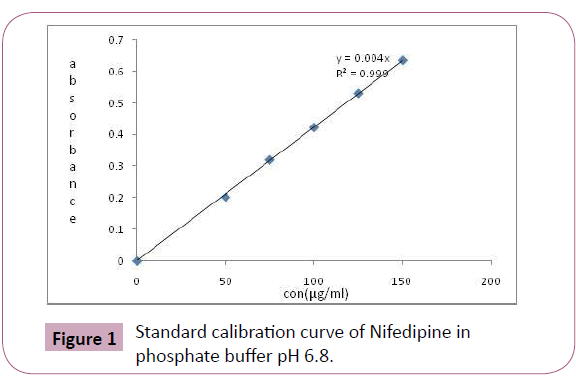
Figure 1: Standard calibration curve of Nifedipine in phosphate buffer pH 6.8.
| TIME (Hrs) |
F1%CDR |
F2%CDR |
F3%CDR |
F4%CDR |
F5%CDR |
F6%CDR |
F7%CDR |
F8%CDR |
F9%CDR |
| 1 |
20.45
±0.51 |
15.0
±0.46 |
16.36±0.31 |
19.77±0.42 |
18.40±0.35 |
14.61±0.58 |
16.70±0.15 |
15.20±0.25 |
14.27±0.12 |
| 2 |
44.65
±0.23 |
26.59
±0.32 |
28.63±0.26 |
42.27±0.23 |
32.04±0.54 |
30.52±0.75 |
31.70±0.50 |
28.63±0.32 |
25.5 ±0.31 |
| 4 |
66.13±0.33 |
42.61±0.62 |
40.22±0.53 |
61.36±0.32 |
50.93±0.34 |
46.06±0.25 |
48.06±0.55 |
45.18±0.63 |
39.36±0.25 |
| 8 |
82.5 ±0.21 |
61.02±0.22 |
53.86±0.23 |
82.84±0.25 |
75.22±0.44 |
62.34±0.65 |
62.04±0.51 |
60.09±0.34 |
57.04±0.63 |
| 12 |
99.20±0.36 |
82.84±0.52 |
73.29±0.65 |
97.84±0.35 |
80.52±0.42 |
80.02±0.51 |
83.86±0.15 |
76.43±0.62 |
74.38±0.32 |
| 16 |
|
98.52±0.24 |
85.56±0.65 |
|
87.84±0.33 |
85.45±0.23 |
98.18±0.32 |
83.52±0.52 |
81.11±0.11 |
| 20 |
|
|
98.18±0.21 |
|
97.84±0.31 |
88.29±0.51 |
|
97.84±0.61 |
95.0 ±0.21 |
| 24 |
|
|
|
|
|
97.18 ±0.23 |
|
|
99.20±0.31 |
Table 5: Results of dissolution profile for F1-F9.
Dissolution profile of formulations F1-F9
The Figure 2 shows the in vitro release profiles of Nefidipine [8] sustained release matrix tablets of formulations F1-F9. Effect of different polymers on the release profile of Nifedipine was studied. In formulations F1, F2, F3 different concentrations of HPMCE5 were used [9]. The release of the drug from the tablet was up to 20 hours only, so these polymers (with in this concentrations) not having the capacity to extend the release up to 24 hours (Table 5). In formulations F4, F5, F6 different concentrations of HPMCK100 were used [10]. The release of the drug from the tablet was up to 24 hours these polymers (with in this concentrations) having the capacity to extend the release up to 24 hours. In formulations F7, F8, F9 different concentrations of Eudragit were used [11]. The release of the drug from the tablet was up to 24 hours these polymers (with in this concentrations) having the capacity to extend the release up to 24 hours.
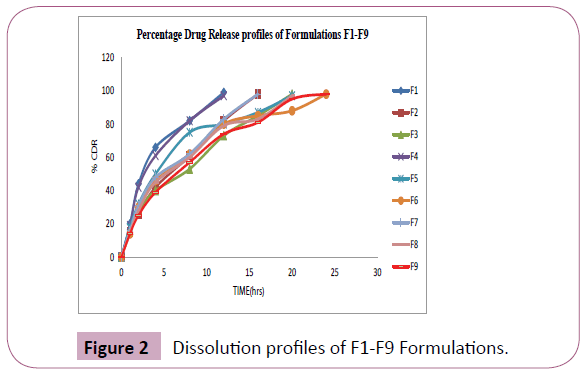
Figure 2: Dissolution profiles of F1-F9 Formulations.
Release kinetics and mechanism
To know the release mechanism and kinetics of Valsartan optimized formulations (F14) were attempted to fit into mathematical models and n, r2 values for zero order [12], First order, Higuchi and Peppas models were represented in Table 6. The peppas model is widely used, when the release mechanism is not well known or more than one type of release could be involved. The semi-empirical equation (Peppas et al.,) [13] shown as equation:
Mt/M∞=k tn
Where, Mt/M∞ is fraction of drug released at time‘t’, k represents a constant, and n is the diffusional exponent, which characterizes the type of release mechanism during the dissolution process (Tables 6 and 7). For non-fickian release, the value of n falls between 0.5 and 1.0; while in case of fickian diffusion, n=0.5; for zero-order release (case II transport), n=1; and for supercase II transport, n > 1
| Formulation code |
Zero Order (R2) |
First Order (R2) |
Higuchi (R2) |
Hixon Crowell (R2) |
KoresmeyerPeppas |
| R2 |
n |
| F1 |
0.873 |
0.9161 |
0.9785 |
0.9771 |
0.9345 |
0.607 |
| F2 |
0.8856 |
0.9683 |
0.9872 |
0.9695 |
0.9967 |
0.664 |
| F3 |
0.9565 |
0.8778 |
0.992 |
0.966 |
0.991 |
0.578 |
| F4 |
0.8927 |
0.9689 |
0.9846 |
0.9902 |
0.9464 |
0.624 |
| F5 |
0.8652 |
0.9526 |
0.9779 |
0.9689 |
0.9673 |
0.544 |
| F6 |
0.8782 |
0.9292 |
0.9816 |
0.9711 |
0.957 |
0.571 |
| F7 |
0.9442 |
0.8917 |
0.9906 |
0.9689 |
0.9802 |
0.5957 |
| F8 |
0.9215 |
0.9211 |
0.9916 |
0.9747 |
0.9803 |
0.5957 |
| F9 |
0.9313 |
0.9498 |
0.9827 |
0.9874 |
0.9887 |
0.4357 |
Table 6: kinetic modeling of F1-F9.
| S.NO |
Time |
log T |
Square root of Time |
%CR |
%Drug remaining |
log %CR |
LOG% DRUG RETAINED |
cube root of %drug remaining |
| 1 |
0 |
0 |
0 |
0 |
100 |
0 |
2 |
4.641589 |
| 2 |
1 |
0 |
1 |
14.27 |
85.73 |
1.154424 |
1.933133 |
4.409381 |
| 3 |
2 |
0.30103 |
1.414214 |
25.5 |
74.5 |
1.40654 |
1.872156 |
4.207771 |
| 4 |
4 |
0.60206 |
2 |
39.36 |
60.64 |
1.595055 |
1.782759 |
3.928738 |
| 5 |
8 |
0.90309 |
2.828427 |
57.04 |
42.96 |
1.75618 |
1.633064 |
3.502311 |
| 6 |
12 |
1.079181 |
3.464102 |
74.38 |
25.62 |
1.871456 |
1.408579 |
2.947993 |
| 7 |
16 |
1.20412 |
4 |
81.11 |
18.89 |
1.909074 |
1.276232 |
2.663242 |
| 8 |
20 |
1.30103 |
4.472136 |
95.03 |
4.97 |
1.977861 |
0.696356 |
1.706549 |
| 9 |
24 |
1.380211 |
4.898979 |
99.2 |
1.8 |
1.992111 |
0.255273 |
1.21644 |
Table 7: kinetic model for optimized formula.
Stability studies
There was no significant change in physical and chemical properties of the tablets of formulation F-9 after 3 Months. Parameters quantified at various time intervals were shown in Table 8.
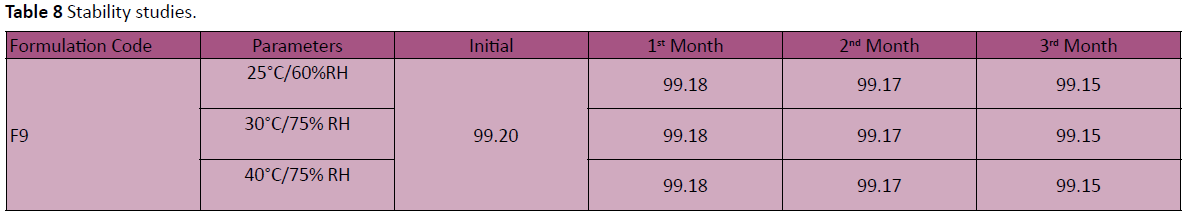
Table 8: Stability studies.
Conclusion
Nifedipine is used in angina. In the present work sustained release tablet was successfully formulated by using different polymers by wet granulation method. The drug-excipient interaction study was carried out using FTIR. In the drug-excipient interaction study, it was found that Nifedipine was having compatibility with all the excipients used in the formulation. Ingredients used are Eudragit, HPMC-E5 and HPMCK100, Lactose, Magnesium Stearate, and Microcrystalline Cellulose
The optimized formula shows,Hardness – 5.7 kgcm2, Friability – 0.04, %CDR – 99.
From the in vitro dissolution analysis the following conclusions are drawn. Formulation batches with HPMC K100 and EUDRAGIT [14] showed better release than the HPMCE5. It was observed that by increasing the viscosity of polymer a retarding effect on the release from the polymer matrix [15,16]. From the dissolution profile modeling it was found that the optimized formulation F9 followed first order release kinetics and Koresmayer Peppas model [17]. Since n>0.5 non Fickian or anomalous diffusion behaviour is generally observed. When the stability results of best formulae was studied at 40°C AND 75% RH for 3 months were compared with their initial results it was found that there was no significant difference in hardness, friability, drug content and drug release of optimized formulation.
5842
References
- Vyas SP, Khar RK (2002) Controlled drug delivery; Concepts and Advances. vallabhprakashan (1stedn). pp. 98.
- Leon S, Andrew YU (1999) Applied biopharmaceutical and Pharmacokinetics. 4th Edition Prentice- Hall International 169-175.
- Afrasim M, Shivakua HG (2010) Formulation of Sustained-release diltiazem matrix tablets using hydrophilic gum blends, Tropical Journal of Pharmaceutical Research 9: 283-291.
- Antesh k, Bhattacharya, pankajverma (2009) Formulation and in vitro evaluation of sustained release matrix tablets of metoprolol succinate using hydrophilic polymers. InternationalJournal of Pharmtech Research 1: 972-977.
- Anton S, Muthu AK, Wagh BP, Manavalan R (2009) Formulation development and evaluation of ondansetron hydrochloride sustained release matrix tablets. J Pharm Sci& Res1: 48-54.
- Bhupendra G,Prajapati, Patel KR (2010) Design and in vitro evaluation of novel nicorandil sustained release matrix tablets based on combination of hydrophilic and hydrophobic matrix system.
- Brahmankar DM, Sunil BJ (2002) Biopharmaceuticsand Pharmacokinetics a Treatise, Vallabhprakashan. 335-337.
- ChittaK, Kishore KR, Ravindra B, Sasidhar V, Abhilash et al. (2010) Designing and evaluation of diclofenac sodium sustained release matrix tablets using hibiscus rosa-sinensis leaves mucilage 1: 29-31.
- Cobby J, Mayersohn M, Walker GC (1974) Influence of shape factors on kinetics of drug release from matrix tablets I Theoretical. J Pharm Sci 725-731.
- Donga JJ, Surani VS, Chauhan SP, Aundhia CJ, Shah NV (2011) Formulation and In-Vitro Evalution of Sustained Release Matrix Tablet of Nifidipine using Hydrophilic Polymer. IJPI’s Journal of Pharmaceutics and Cosmetology1:10-16.
- Ganesh R. Suresh K, Jawahar, Senthil V, Nagasamy D, et al. (2010) Preparation and evaluation of sustained release matrix tablet of diclofenac sodium using natural polymer. Journal of Pharmaceuical science & Research2: 360-368.
- Harekrishna R, Anup C, Bhabani SN, Satyabrata B, Sruti RM (2010) Design and in vitro evaluation of sustained release matrix tablets of complexednicardipine hydrochloride. International journal of pharmacy and pharmaceutical sciences 2: 128-132.
- Herbert A, Lieberman, Leon L (1989) Pharmaceutical Dosage Forms, Tablets, Marcel Dekker Inc., New York, 3: 9-10.
- HosseinaliT, Seyed AM, Tina BG (2003) Preparation of sustained-Release Matrix Tablets of Aspirin with Ethylcellulose, Eudragit RS100 and Eudragit S100 and Studying the Release Profiles and their Sensitivity to Tablet Hardness. Iranian Journal of Pharmaceutical Research 201-206.
- Singh K, Kumar A, Langyan N, Ahuja M (2009) Evaluation of mimosa pudica seed mucilage as sustained-release excipient. AAPS PharmSciTech 10: 1121-1127.
- Peppas NA (1985) Analysis of Fickian and non-Fickian drug release from polymers. Pharm ActaHelv 60: 110-111.
- Siepmann J, Kranz H, Bodmeier R, Peppas NA (1999) HPMC-matrices for controlled drug delivery: a new model combining diffusion, swelling, and dissolution mechanisms and predicting the release kinetics. Pharm Res 16: 1748-1756.