Keywords
|
| Dementia; Psychiatric disorders; Proteomics |
Introduction
|
| In the last years, “omics” technologies have had a great impact on the scientific community and disciplines like Genomics, i.e. the study of the complete set of DNA within a single cell of an organism, and Proteomics, i.e. the study of all the proteins produced and modified by an organism, have markedly spread. |
| A recent employment of these technologies is in the research of new biomarkers for the early detection of brain diseases. In this quest, it could be determinant the observation that energy availability and exploitation are fundamental to grant neuronal functions and survival, because any alterations in energy metabolism affect proper physiology. By the way, many studies indicate that changes in brain energy metabolism are causative of development/progression of several diseases, only subsequently implying morphological modifications [1-5]. Remarkably, this last statement is valid both for neurological and, more recently, for psychiatric disorders, for which the term “Mitochondrial Psychiatry” has been coined [6]. In any case, the paradigmatic example to describe the role of brain energy metabolism as a key etiopathological factor of brain disease and biomarker discovery is Dementia. |
| The World Health Organization (WHO) estimates that 35.6 million people live with Dementia, a number that is anticipated to triple by 2050. Epidemiological, clinical and neuropathological studies in Europe, North America and Asia showed that, out of 6-7% of cases of dementia in the population over 65 years, Vascular Dementia (VAD) accounts for 15 to 30% and Dementia or Dementia of Alzheimer’s type (AD) from 50 to 70%. |
| However, there are also many “Mixed Dementia” cases, better classified as “AD with CerebroVascular Disease”, in which variable proportions of vascular and degenerative lesions (peculiar of AD) coexist [7]. Overlap of AD neuropathology (amyloid plaques and neurofibrillary tangles) with cerebrovascular lesions is observed in up to 50% of cases of Dementia [8]. These lesions include atherosclerosis of the circle of Willis and its branches, leukoaraiosis and lacunar infarcts, microbleeds, microinfarcts and cerebral amyloid angiopathy [8-11]. |
| Moreover, AD and VAD share many genetic and environmental risk factors, such as ApoE allele E4, Coenzyme Q, Vitamin E [12] diabetes and hyperinsulinemia, high systolic blood pressure in middle and late ages, low diastolic blood pressure in late ages, smoking, previous stroke and head trauma, high homocysteine serum levels, hypercolesterolemia and atherosclerosis [13]. |
| Thus, the overall effect on cognition would result from the combined burden of vascular and neurodegenerative pathology, according to an additive model. Alternatively, vascular disease could promote AD and vice-versa, resulting in reciprocal interactions amplifying their pathogenic effects [14]. Remarkably, several studies [15-17] suggested that, in older individuals with moderate AD pathology, subcortical vascular lesions are the major determinants of the expression of Dementia. For the clinical practice and therapy, these considerations support the absolute need of increasing the understanding of (i) the impact of vascular factors and cerebrovascular disease on AD, and (ii) of the neurodegenerative processes of AD on VAD. Accordingly, the development of biomarkers of key microvascular/cellular processes may help to identify these complex interactions between AD and VAD, particularly in the early stages of the disease. |
| In this perspective, it has been decisive starting to consider the tight relationship between cerebrovascular disease, cerebral blood flow and brain energy metabolism: this concept firstly appeared in 1970s and was called micro-vascular-tissutal unit [18]; now is known as neurovascular unit and has allowed to shift the focus of the problem from the brain vessels to intracellular molecular modifications linked to brain Bioenergetics. |
| At this regard, impaired energy metabolism has been detected also directly in patients, where reductions of cerebral metabolic rate for glucose (CMRglu), cerebral metabolic rate for O2 (CMRO2) and of regional cerebral blood flow (rCBF) was observed to be critically involved in cognitive decline and Dementia [7], as respectively shown by imaging assessment with PET or MRI [19] and by glucose uptake of 18F-2-fluoro-2- deoxy-D-glucose [20]. In humans, electron microscopic studies showed: (i) accumulation of abnormal mitochondria in AD dystrophic neurites [21]; (ii) abnormal mitochondria impoverished in cytochrome c oxidase (Complex IV of the Electron Transfer Chain), which activity is decreased in AD brains [22]; (iii) degenerated mitochondria in dystrophic neurites in aged Rhesus monkeys [23]. Reduced O2 uptake, glycolysis and Krebs’ cycle have been also recognized in AD and in pre-clinical stages in APP23 mouse model [24]. Moreover, human brain energy metabolism impairments were detected in Dementia [25,26], in familial AD by neuroimaging studies, before showing clinical signs of atrophy detected by MRI evidence [27]. |
| Although these deficiencies may be the result of cell death and neuronal loss in advanced stages of the disease, the presence of similar abnormalites in pre-clinical stages underline the importance of impaired energy metabolism during aging as causative, and consequently should it be considered as the major determinant for the development of AD clinical evidence and for drug treatment efficacy. The temporal relationship of reductions in CMR prior to the development of clinical disabilities is consistent with the fact that CMR reductions happen before brain lesions become apparent, and subtle changes may accompany the aging process even in unimpaired but aged individuals [2,3,28]. A three metabolic homeostatic/dismetabolic dynamic concept model far from steady-state, was hypothesized by us respect to the entire time course of life during aging [2]. |
| The pathophysiological consequence of the perturbed energy metabolism and transduction processes in demented individuals is the fall of ATP production in mitochondria, by around 50% in the beginning of AD, increasing during its course [29]. The ATP fall induces not only lipid peroxidation but also, more importantly, the formation of peroxynitrites affecting the functionality of structural and catalytic proteins (i.e. enzymes) in mitochondria for the synthesis of ATP, in synaptic plasma membranes for the utilization of ATP by neurons and glial cells. The concequence is the impaired functional Proteomics of the associated enzyme activities (e.g. acetylcholinesterase), of the glucose phosphorylation for its transport from blood to neurons (e.g. hexokinase) and of the ion-motive and synaptic homeostasis and neurotransmission (e.g. ATP-ases, whose functionality is already dampened by the ATP deficit itself) [30]. All these catalytic dysfunctions in neuronal cells are obviously linked and above all integrated between each other, leading to the concept of functional Metabolomics. |
| These concepts are exemplified in Figure 1, that is an abstract picture of this functional brain network, where it is highlighted the role of functional Proteomics as a useful technology to study the neuronal biochimism, allowing to evaluate the metabolic changes within neuronal cells and brain tissue that may imply some physiopathological modifications. A key feature of this type of approach is that these metabolic changes are considered in their completeness and as a whole, i.e. evaluating the functional Metabolomics. Moreover, respect to Proteomics, that describes the concentrations of proteins in a given time frame, functional Proteomics allows to evaluate the functional potentialities of cells. |
| These considerations underline that age-related changes in brain energy metabolism and in mitochondrial functionality should be considered as remarkable factors during physiological aging and related pathophysiogical events, above all Dementias and AD and they may be of main importance also as molecular targets for pharmacological treatments [1-3,28,30-36]. |
| Given these premises, the critical barrier that have dampened so far the understanding of the vascular/metabolic contributions to AD is the lack of studies systematically linking vascular factors, brain energy metabolism, AD pathology progression and responsiveness to pharmacological treatment. In this perspective, methodical investigations assessing the enzyme activities of the Krebs’ Cycle, Electron Transport Chain complexes and energy-consuming systems of ATPases may be decisive in overcoming this limit, particularly if this studies are conducted on subcellular fractions. Current technologies [2,28,35,36] make these studies possible both in samples of cerebral tissue from animals and of humans. In fact, many peripheral vascular cells of AD and VAD patients, like lymphocytes, show several molecular alterations, including mitochondrial dysfunction, that mirror those occurring in the brain of demented patients when evaluated post exitus, as preliminarily shown by us [12,37] and by Literature [38]. Therefore, lymphocytes could be studied for biomarkers development to assess if energy-linked enzyme activities could be predictive markers of neuronal energy transduction abnormalities, linking vascular and AD pathology. |
Figures at a glance
|
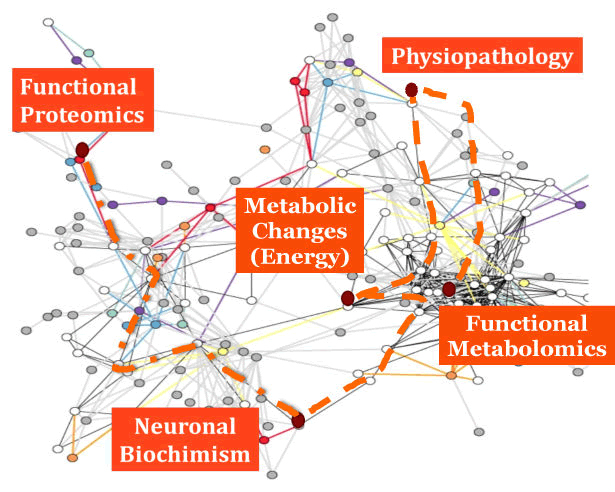 |
| Figure 1 |
|
| |
|
|

