Key words
Autoimmunity. Major Histocompatibility complex. Epidemiology. Multiple sclerosis. Molecular genetics.
La esclerosis múltiple (EM) es la enfermedad inflamatoria crónica más frecuente del sistema nervioso central (SNC), con una alta incidencia en países del Norte de Europa [1], pero que afecta personas en todo el mundo. En años recientes, y cada vez con mayor frecuencia, ha sido reportada en Latinoamérica [2]. Estudios epidemiológicos se han llevado a cabo en Colombia, demostrando una prevalencia de 1,48 a 4,98 por cada 100.000 habitantes [3]. En la ciudad de Medellín (Colombia), se han adelantado estudios genéticos bien diseñados que han mostrado susceptibilidad genética que predispone al desarrollo de la enfermedad [4,5]
La EM se caracteriza por desmielinización, inflamación y gliosis [6,7], cuya presentación clínica se clasifica como primaria, secundariamente progresiva o recaída-remisión [8]. Las lesiones de la EM son típicamente diseminadas; su cuadro clínico puede variar desde un trastorno benigno autolimitado hasta una enfermedad severa, prolongada y altamente incapacitante [9].
Aunque esta enfermedad afecta principalmente a adultos jóvenes, también ha sido bien demostrado por estudios clínicos y patológicos, que la EM puede comenzar en la infancia, aunque su prevalencia en este grupo de edad es mucho menor que en la población adulta [10-12].
La patogénesis de la EM no ha sido completamente dilucidada, pero la información existente apunta a que ésta es una enfermedad multifactorial en la que participan factores genéticos e inmunológicos y factores ambientales disparadores [13]. En este sentido, varias teorías han sido propuestas como eventos desencadenantes del proceso autoinmune que conlleva a los cambios patológicos propios de la enfermedad, incluso –como lo sugiere un estudio español realizado por Alemany-Rodríguez et al- en asociación con otras enfermedades ya definidas como autoinmunes [14,15]. La más probable hace referencia a la existencia de una infección viral, en la cual existen autoantígenos que generan mimetismo molecular con proteínas de la mielina, ocasionando una pérdida de tolerancia contra éstos, lo cual deriva en la destrucción de la mielina mediada por linfocitos T activados [16-21]. Sin embargo, para que esto ocurra, es necesaria la presencia de una susceptibilidad genética en el individuo, la cual es sugerida por varias observaciones tales como la diferente prevalencia entre grupos étnicos que habitan una misma región [22] y la mayor prevalencia de la enfermedad en gemelos monocigóticos que en dicigóticos [23]. Aunque algunos casos familiares de EM han sido reportados, esta enfermedad esta lejos de ser explicada por una herencia mendeliana simple, y su complejidad está en relación con la participación de los diferentes factores antes mencionados.
Existe gran dificultad para discriminar la real influencia de lo genético y lo ambiental en la aparición de la EM, a este respecto un estudio colombiano fue realizado en una población perteneciente al trópico, logrando demostrar que un factor genético (en este caso el desequilibrio del ligamiento al HLA DQα) estaba presente, al igual que en poblaciones caucasoides no tropicales, revelando así más argumentos para pensar en la participación del componente genético como disparador de la enfermedad [24].
Adicionalmente, algunos autores españoles, como Fernández y Uría, en diferentes publicaciones, han propuesto que el componente genético resulta insuficiente para explicar la presentación de la enfermedad, y sugieren que ambos factores (genético y ambiental) interactúan y potencian el efecto biológico y molecular que deriva en el desarrollo de la EM. A este respecto, se mencionan argumentos, no necesariamente excluyentes, de cada una de las hipótesis; las más importantes son, para el factor ambiental, la prevalencia variable en diferentes lugares del mundo, cambios en la incidencia en cortos períodos de tiempo y descripciones en la literatura de posibles focos y epidemias (p. ej., Islas Feroe, Islandia, Kenia, Malta, entre otros). Del factor genético se menciona, entre otros, la presencia de grupos étnicos ¨resistentes¨ y ¨susceptibles¨, asociación con el HLA, recurrencia entre hermanos y predominio en gemelos monocigóticos sobre los dicigóticos [25,26].
Objetivo
Describir los aspectos genéticos relevantes sobre la EM, producto del análisis de datos epidemiológicos y moleculares, con base en la literatura médica mundial publicada hasta la actualidad, proporcionando nuevas posibilidades en el entendimiento de la patogénesis de esta enfermedad y opciones terapéuticas potenciales para los pacientes que la padecen.
Desarrollo Del Tema
Epidemiología Genética
Estudios familiares
La agregación familiar de la esclerosis múltiple fue primero descrita por Charcot a finales del siglo XIX [27], pero sólo hasta el siglo XX se adelantaron estudios poblacionales sistemáticos [28-31].
En la década de los años sesenta, MacKay et al., encontraron que el riesgo de desarrollar EM es 20 veces mayor en familiares de primer grado de consanguinidad de pacientes con EM que en la población general [32]. Estos hallazgos fueron luego corroborados por Sadovnick et al., en 1988 [33], quienes desarrollaron un estudio sistemático ajustado por edad, encontrando mayor riesgo de la enfermedad en familiares de primero, segundo y tercer grado de consanguinidad. Estos autores demostraron cómo el riesgo es mayor a medida que la relación es más cercana. Mas recientemente, Willer et al., [23] desarrollaron un estudio poblacional de 370 casos de EM en Canadá, encontrando una concordancia del 25,3% en gemelos monocigóticos, 5,4% en dicigóticos y 2,9% en hermanos no gemelos. A pesar de lo anterior, es el común denominador de todos los estudios hasta la fecha publicados, que no se pueda establecer un patrón de herencia lógico, lo anterior corroborado por un estudio ibérico en seis familias con al menos un caso de EM, en donde no se logra establecer un patrón de herencia determinado [34].
Si bien la agregación familiar de la EM había sido estimada alrededor del 20% en estudios previos [30,31], el análisis de dos poblaciones diferentes en Hungría [35] y en Irán [36], que fueron publicados recientemente, sugieren que este porcentaje puede ser menor, ubicándose alrededor del 10%. En España, Fernandez-Pérez et al., reportan una prevalencia de EM familiar incluso menor a las citadas, con un 7,47% en la población objeto de su estudio [37].
La tabla 1 muestra el riesgo de aparición de la enfermedad según la relación con el caso índice estimado en estudios poblacionales ajustados por edad [38-40].

Tabla 1. Riesgo de EM según parentesco con caso índice
No obstante, los estudios poblacionales demuestran claramente la asociación familiar de la enfermedad, pero, como se menciono previamente, no responden a un modelo de herencia mendeliana simple y, por el contrario, hacen pensar que ésta es una enfermedad en la que participan varios genes con una transmisión familiar compleja.
Estudios de adopciones
Cuando se analiza la agregación familiar de una enfermedad siempre existe se duda si ésta es debida a la herencia o a la influencia del ambiente familiar compartido. Para esto se compara el riesgo de recurrencia de individuos adoptados –sin relación de consanguinidad con la familia que conviven– con respecto a miembros naturales de la misma familia que comparten material genético. Con respecto a esto, los estudios no han demostrado que estos individuos adoptados tengan mayor riesgo de desarrollar EM que la población general [41], lo que soporta la idea de que son precisamente factores genéticos los que determinan la agregación familiar de la EM por encima de los factores medioambientales.
En este sentido, el grupo canadiense colaborativo para el estudio de la EM, hace más de 10 años, analizó una población de 15.000 pacientes con EM en búsqueda de casos índices adoptados o de aquellos pacientes que convivían con individuos adoptados o hijos no naturales. Un total de 1.201 individuos no naturales convivían con los casos índice (470 Padres, 345 hermanos y 386 hijos), de los cuales sólo uno resultó afectado por la enfermedad, lo cual corresponde a la misma prevalencia en la población general [41].
Estudios en hermanos medios
Los estudios en hermanos medios –que sólo comparten un progenitor biológico– permiten identificar la influencia específica de cada padre en la transmisión de la enfermedad. Ebers et al., compararon el riesgo de recurrencia en hermanos medios con respecto al padre que tienen en común, para lo cual analizaron 1.567 casos de pacientes con EM quienes tenían 3.436 hermanos medios. El riesgo global para los hermanos medios fue del 1,89% comparado con el 3,11% de los hermanos completos. Pero al discriminar por padre en común se encontró que para la madre el riesgo es del 2,35% comparado con el 1,31% de riesgo paterno (p=0,048), lo cual sugiere un efecto de origen materno que aumenta la susceptibilidad de sufrir la enfermedad comparada con el riesgo de origen paterno [42]. Estos hallazgos difieren de los encontrados en un estudio previo publicado por el mismo grupo donde no encontraron diferencia entre padres en común [43]; estudios adicionales son necesarios.
Se ha sugerido que la EM es mucho más frecuente en mujeres; en este contexto, si los hombres tienen mayor resistencia biológica a la enfermedad, necesitarían una predisposición genética mayor para poder desarrollarla; si esto fuese cierto, los hombres transmitirían la enfermedad más frecuentemente que las mujeres, fenómeno conocido como Efecto Carter. Una publicación reciente del grupo de la Clínica Mayo y la Universidad de California, demuestra este “efecto” en 197 familias estudiadas [44].
Estudios en gemelos
La forma clásica para diferenciar la influencia de los genes y del ambiente son los estudios en gemelos, muchos de los cuales se han adelantado en pacientes con EM. Como se mencionó previamente, el riesgo de desarrollar la enfermedad para gemelos monocigóticos se ha estimado cercano al 30%, mientras que en dicigóticos se aproxima al 5% [23,40,45-48]. No obstante, estudios realizados en diferentes poblaciones demuestran cómo estas proporciones pueden cambiar dependiendo de la población estudiada. Ristori et al., analizaron dos poblaciones diferentes en Italia continental y Cerdeña (isla italiana ubicada sobre el mediterráneo), encontrando que para los continentales el riesgo de los monocigóticos era del 14,5%, y de los heterocigóticos del 4%, mientras que para los isleños el riesgo de los monocigóticos era del 22,2% y 0% para los dicigóticos [49]. Estas observaciones apuntan a que la interrelación entre factores genéticos y ambientales es compleja y varía de acuerdo a la ubicación geográfica de la población.
Para añadir aún mas discusión a este debate entre genes y ambiente, en un artículo de publicación recientemente se estudió la diferencia de exposición al sol entre gemelos monocigóticos discordantes para la enfermedad, encontrando que aquellos que se exponían menos al sol tenían mayor riesgo de desarrollar la enfermedad o, dicho en otras palabras, la exposición al sol protege contra el desarrollo de la enfermedad independientemente de la susceptibilidad genética (OR de 0,4, IC 95%: 0,19 – 0,83) [50].
Estudios en hijos de ambos padres con EM
Varios estudios poblacionales han sido desarrollados para calcular el riesgo de EM cuando ambos padres tienen la enfermedad. Estos han mostrado que los hijos de estas parejas tienen un riesgo similar o superior al de los gemelos monocigóticos (alrededor del 30%) comparado con el 2,49% cuando sólo uno de los padres es afectado [51,52]. En un estudio reciente realizado en Irán se encontró que casarse con un paciente con EM puede aumentar el riesgo de padecer la enfermedad hasta 12,5 veces. Estos autores analizaron los 1.076 pacientes con EM que estaban casados en dicho país, encontrando que seis de éstas parejas desarrollaron la enfermedad después del matrimonio, lo que representa un riesgo del 0,5% comparado con el 0,04% de la población general [53]. Este porcentaje, si bien no es muy alto, indica la participación de un ambiente común más que el de una infección de transmisión sexual [54].
Genética Molecular
Varios genes han sido propuestos como posibles candidatos en la génesis de la EM, aunque ninguno de ellos de forma concluyente. A continuación se presentan los que cuentan con mayor evidencia.
Complejo Mayor de Histocompatibilidad (CMH)
Las moléculas del CMH o también llamado antígeno leucocitario humano (HLA) ayudan a diferenciar los antígenos propios de los antígenos extraños, por lo que estas moléculas han sido fuertemente asociadas con la aparición de diferentes enfermedades autoinmunes [55].
En distintos grupos étnicos se ha encontrado asociación entre EM y alelos específicos del antígeno leucocitario humano (HLA), especialmente con el HLA clase II [56]. Se ha demostrado que el alelo HLA-DRB1*15 incrementa dos a tres veces el riesgo de desarrollar EM [56,57-59], especialmente una variante inmunogenética que cursa con bandas oligoclonales en el líquido cefalorraquídeo, mientras que el alelo HLA DRB1*04 se ha asociado a una variante con bandas oligoclonales negativas en el líquido cefalorraquídeo [60]. También existen algunos estudios que han asociado otros alelos DR con la predisposición para desarrollar la enfermedad, específicamente el HLA-DRB1*17 [61]. Por el contrario, existe evidencia de que algunos alelos HLA son protectores para el desarrollo de la enfermedad; tal es el caso del HLA-DRB1*14 [61,62]. Estudios realizados en Medellín, Colombia, han demostrado que polimorfismos del gen HLA DQA1 ubicado en el cromosoma 6p21.3-21.4, se asocian a mayor o menor predisposición para desarrollar EM [4,5].
El mecanismo por el cual estas moléculas favorecen el desarrollo de EM ha sido estudiado en modelo murino con expresión transgénica del HLA-DRB1*15, desarrollando inmunidad contra la proteína básica de la mielina (PBM) y un síndrome clínico similar a la EM [63]. Además, en ratones con expresión transgénica del mismo gen se desarrolló inmunidad contra otro autoantígeno, la glicoproteína oligodendrocítica de la mielina (GOM), con posterior desmielinización del SNC [64].
En España se recogió la evidencia disponible con respecto a la asociación entre HLA y EM, encontrando que las formas primariamente progresivas y las de peor pronóstico se asociaron a DR4 y las formas benignas a DR2. El DRw13 (DR6) aparece como un posible alelo protector [65].
Estudios genéticos moleculares, gravedad clínica de la enfermedad y asociación con el sexo
Numerosos estudios han sido publicados intentando establecer una relación directa entre un gen específico y la gravedad de la EM. En población hispanoamericana existe una publicación española que encontró asociación entre la presencia de DRB1*1501 y categorías bajas en la escala EDSS (del inglés, expanded disability status scale), sin embargo esta asociación fue exclusiva de los hombres, ya que en las mujeres se encontró asociación entre la presencia de este gen con el desarrollo de la enfermedad, pero no se logró demostrar una relación directa con la gravedad de la misma [66]. Por su parte, De la Concha et al., en un estudio habían determinado que la asociación de formas benignas de la enfermedad se daba sólo en mujeres, al mismo tiempo que atribuyen al DRB1*1501 exclusividad de presentación en ellas [67]. A este respecto, los autores del estudio español previamente mencionado, alegan que es incoherente que un factor ¨promueva¨ la presentación de una enfermedad y al mismo tiempo la haga benigna. [66]. A pesar de la objeción anterior, es conocido el modelo etiológico en diversas enfermedades, en el cual algunos factores causales poseen un menor índice de patogenicidad y, por tanto, menor capacidad de lograr cambios fenotípicos mayores.
Polimorfismos del gen del receptor de interleucina 7 alfa (IL7RA) e interleucina 2 alfa (IL2RA)
Estudios de mapeo genético en humanos han establecido una relación indiscutible entre los genes IL7RA e IL2RA y la EM, éste es, quizá, el avance más grande en la genética de la esclerosis múltiple en los últimos años [68,69].
Recientemente fue publicado un estudio de asociación en todo el genoma humano para identificar alelos relacionados con el riesgo de EM. En este estudio con más de 12.000 participantes se encontró que los alelos de los genes IL2RA e IL7RA son factores de riesgo heredables para el desarrollo de la enfermedad [70].
Otros genes relacionados
Polimorfismos del receptor beta de células T son potenciales candidatos para susceptibilidad en EM, sin embargo la evidencia que soporta esta asociación no es clara; al parecer estos polimorfismos actúan epistáticamente con genes relacionados del HLA para aumentar la susceptibilidad [71-73].
Estudios realizados en la década de los noventa arrojaron algunos hallazgos positivos en relación con polimorfismos del gen que codifica para la PBM (ubicado en el cromosoma 18q22) y la susceptibilidad para la EM. Tienari et al., realizaron un estudio de casos y controles en una población de Finlandia donde existe una fuerte agregación familiar de la enfermedad, encontrando una alta prevalencia del alelo de 1,27 Kb del gen de la PBM en los pacientes afectados, con significancia estadística (p=0,000049) [74]. Estos hallazgos no fueron confirmados en estudios posteriores [75,76].
Otros genes han sido estudiados como posibles candidatos en la susceptibilidad para la EM, pero los hallazgos no han sido concluyentes; hace falta investigación al respecto. En la tabla 2 se nombran algunos de ellos.
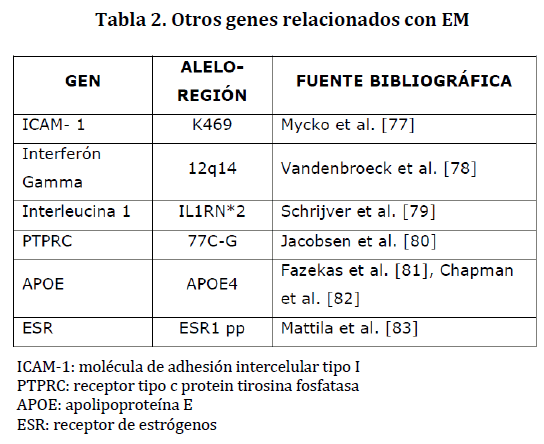
Tabla 2. Otros genes relacionados con EM
Conclusión y Perspectivas
• La EM es una enfermedad compleja cuya aparición está determinada por la interrelación entre factores genéticos y ambientales.
• Existe amplia evidencia que documenta la susceptibilidad genética de la enfermedad, pero faltan estudios que aporten en la identificación de genes específicos de peso en la génesis de la EM.
• No es ésta una enfermedad monogénica y son muchos los genes que participan en la susceptibilidad para desarrollar este trastorno. Los avances en los estudios de genética molecular que se alcancen en años venideros serán de suma importancia en la identificación de genes específicos y en la implementación de nuevos tratamientos enfocados en blancos moleculares, que podrán, no sólo modificar el curso de la enfermedad, sino también prevenir su aparición en los individuos con riesgo familiar.
• No existe consenso sobre el predominio de influencia etiológica entre el factor genético y el ambiental, lo cual sugiere que, con evidencia de ambas hipótesis, la enfermedad esté determinada por un factor genético que requiera un desencadenante ambiental. Estudios adicionales al respecto son necesarios y deben enfocarse a evaluar la posibilidad de factores concomitantes, no necesariamente excluyentes, interviniendo en el desarrollo de la EM.
• En la actualidad se encuentra en fase 2 de investigación una vacuna DNA (BHT- 3009) que codifica para la proteína básica de la mielina, induciendo tolerancia inmunológica antígeno-específica. Esta vacuna demostró reducción en la aparición de nuevas lesiones en la resonancia magnética de pacientes con EM en recaída-remisión [84]. Este hallazgo genera opciones a corto plazo para los pacientes que padecen esta enfermedad y aportan futuras posibilidades de investigación.
979
References
- Corona T, Roman GC. Multiple Sclerosis in Latin America. Neuroepidemiology 2006; 26(1): 1-3.
- Sánchez JL, Aguirre C, Arcos OM, Jiménez I, Jiménez M, León F, et. Al. Prevalencia de la esclerosis múltiple en Colombia. Rev Neurol 2000; 31: 1101–3.
- Palacio LG, Rivera D, Builes JJ, Jiménez ME, Salgar M, Anaya JM, Jiménez I, Camargo M, Arcos-Burgos M, Sánchez JL. Multiple sclerosis in the tropics: genetic association to STR’s loci spanning the HLA and TNF. Mult Scler 2002; 8: 249–55.
- Arcos-Burgos M, Palacio G, Sánchez JL, Londoño AC, Uribe CS, Jiménez M, Villa A, Anaya JM, Bravo ML, Jaramillo N, Espinal C, Builes JJ, Moreno M, Jiménez I. Multiple sclerosis: association to HLA DQ in a tropical population. Exp Clin Immunogenet 1999; 16: 131–8
- Lassmann H. The pathology of multiple sclerosis and its evolution. Phil Trans R Soc Lond B 1999; 354: 1635-40
- G. Martino, R. Furlan, P.L. Poliani. El significado patogénico de la inflamación en la esclerosis múltiple. Rev Neurol 2000; 30 (12): 1213-7.
- Kantarci OH, Weinshenker BG. Natural history of multiple sclerosis. Neurol Clin 2005; 23: 17-38.
- Heard RN. The spectrum of multiple sclerosis. Curr Allergy Asthma Rep 2007; 7(4): 280-4.
- Chitnis T. Pediatric multiple sclerosis. Neurologist 2006; 12(6): 299-310.
- Boiko A, Vorobeychik G, Paty D, Devonshire V, Sadovnick D et al. Early onset multiple sclerosis. A longitudinal study. Neurology 2002; 59: 1006-10.
- Shaw CM, Alvord EC, Jr. Multiple sclerosis beginning in infancy. J Child Neurol 1987; 2: 252–6.
- Sawcer S. The complex genetics of multiple sclerosis: pitfalland prospects. Brain 2008, May: 1-14.
- Grigoriadis N, Grigoriadis S, Polyzoidou E, Milonas I, Karussis D. Neuroinflammation in multiple sclerosis: Evidence for autoimmune dysregulation, not simple autoimmune reaction. Clin Neurol Neurosurg 2006; 108: 241-4.
- M. J. Alemany-Rodríguez, Y. Aladro, R. Amela-Peris, M. C. Pérez-Viéitez, M. P. Reyes Yañez, M. C. Déniz-Naranjo. Rev Neurol 2005; 40 (10): 594-7.
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995; 80: 695-705.
- Correale J, Tenembaum S. Myelin basic protein and myelin oligodendrocyte glycoprotein T-cell repertoire in chidhood and juvenile multiple sclerosis. Multiple sclerosis 2006; 12: 1-9.
- Minagar A, Carpenter A, Alecander JS. The destructive alliance: interactions of leukocytes, cerebral endothelial cells, and the immune cascade in pathogenesis of multiple sclerosis. Int Rev Neurobiol 2007; 79: 1-11.
- Lunemann JD, Munz C. Epstein-Barr virus and multiple sclerosis. Curr Neurol Neurosci Rep. 2007; 7(3): 253-8.
- Liu X, Lee YS, Yu CR, Egwaugu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Inmunol 2008 May 1; 180(9): 6070-6.
- Alvarez-Lafuente R, et al. Herpesviruses and human endogenous retroviral sequences in the cerebrospinal fluid of multiple sclerosis patients. Mult Scler 2008; 14 (5): 595-601.
- Alter M, Kahana E, Zilber N, Millar A. Multiple sclerosis frequency in Israel's diverse populations. Neurology 2006; 66(7): 1061-6.
- Willer CJ, Dyment DA, Sadovnick AD, et al. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci (USA) 2003; 100: 12877–2.
- J. L. Sánchez, L. G. Palacio, A. C. Londoño, C. S. Uribe, M. E. Jiménez, L. A. Villa, et al. Esclerosis múltiple: aproximación epidemiológico-genética en habitantes de Antioquia, Colombia. Desequilibrio del ligamiento al HLA DQα. Rev Neurol 2000; 30 (2): 170-3.
- O. Fernández. Factores genéticos y ambientales en la esclerosis múltiple. Rev Neurol 2000; 30 (10): 964-7.
- D. F. Uría. HLA Epidemiología genética de la esclerosis múltiple. Rev Neurol 2002; 35 (10): 979-84.
- Charcot JM. Oeuvres Completes. Lecons sur les Maladirs du Systeme Nerveux. Paris: Tome I; 1894: 269.
- Pratt RTC. The familial occurrence of disseminated sclerosis. Ann Eugen (Lond). 1951; 16: 45–9.
- Pratt RTC, Compston ND, McAlpine D. Familial incidence of disseminated sclerosis and its significance. Brain 1951; 74: 191–32.
- Mackey RP. The familial occurrence of multiple sclerosis and its implications. Res Publ Assoc Res Nerv Ment Dis 1950; 28: 150–77.
- Sadovnick AD, MacLeod PMJ. Familial nature of multiple sclerosis: empiric risk for first, second and third degree relatives of patients. Neurology 1981; 31: 1039–41.
- MacKay RP, Myrianthopoulos NC. Multiple sclerosis in twins and their relatives: final report. Arch. Neurol 1966; 15: 449-62.
- Sadovnick AD, Baird PA, Ward RH. Multiple Sclerosis: updated risks for relatives. Am J Med Genet 1988; 29: 533–41.
- L. Landete, B. Casanova, J. A. Burguera. Esclerosis múltiple familiar: estudio de seis familias. Rev Neurol 1998; 27 (155): 43-7.
- Fricska-Nagy Z, Bencsik K, Rajda C, Fuvesi J, Honti V, Csepany T, Dobos E, Matyas K, Rozsa C, Komoly S, Vecsei L. Epidemiology of familial multiple sclerosis in Hungary. Mult Scler 2007 Mar; 13(2): 260-1.
- Saadatnia M, Etemadifar M, Maghzi AH. Multiple sclerosis in Isfahan, Iran. Int Rev Neurobiol 2007; 79: 357-75.
- M. J. Fernández-Pérez, O. Barakat, J. M. García-Moreno, M. Lucas, G. Navarro, J. M. Gata, et al. Características clínicas de la esclerosis múltiple en España. Rev Neurol 1999; 29 (8): 693-6.
- Robertson N, Fraser Deans J, Clayton D, Walker N, Compston D. Age-adjusted recurrence risks for relatives of patients with multiple sclerosis. Brain 1996; 119: 449–5.
- Carton H, Vlietinck R, Debruyne J, Dekeyzer J, D’Hooghe M, Medaer R, Truyen L, Sadovnick AD. Recurrence risk of MS in relatives of patients in Flanders, Belgium. J Neuroimmunol 1995; S(1): 54.
- Sadovnick AD, Armstrong H, Rice GP, Bulman D, Hashimoto L, Paty DW, Hashimoto SA, Warren S, Hader W, Murray TJ, Seland TP, Mety L, Bell R, Duquette P, Gray T, Nelson R, Weinshenker B, Brunet D, Ebers GC. A population-based study of twins: Update. Ann Neurol 1993; 33: 281–5.
- Ebers GC, Sadovnick AD, Risch NJ, and the Canadian Collaborative Study Group. A genetic basis for familial aggregation in multiple sclerosis. Nature 1995; 377: 150–1.
- Ebers GC, Sadovnick AD, Dyment DA, Yee IM, Willer CJ, Risch N. Parent-of-origin effect in multiple sclerosis: observations in half-siblings. Lancet 2004; 363(9423): 1773-4.
- Sadovnick, AD, Ebers GC, Dyment DA, Risch NJ. Canadian Collaborative Study Group: Evidence for genetic basis of multiple sclerosis. Lancet 1996; 347: 1728-30.
- Kantarci OH, Bracellos LF, Atkinson EJ, Ramsay P et al. Men transmit MS more often to their children vs women. The Carter effect. Neurology 2006; (67): 305-10.
- Bobowick AR, Kurtske JF, Brody JA, Hrubec Z, Gillespie M. Twin study of multiple sclerosis: an epidemiological inquiry. Neurology 1978; 28: 978–7.
- Ebers GC, Bulman DE, Sadovnick AD, et al. A population-based study of multiple sclerosis in twins. New Engl J Med 1986; 315: 1638–2.
- Kinnumen E, Koskenvuo M, Kaprio J, Aho K. Multiple sclerosis in a nationwide series of twins. Neurology 1987; 37: 1627–9.
- Hansen T, Skytthe A, Stenager E, Petersen HC, Brønnum-Hansen H, Kyvik KO. Concordance for multiple sclerosis in Danish twins: an update of a nationwide study. Mult Scler 2005; 11(5): 504-10.
- Ristori G, Cannoni S, Stazi MA, Vanacore N, Cotichini R, Alfo M, Pugliatti M, Sotgiu S, Solaro C, Bomprezzi R, Di Giovanni S, Talamanca LF, et al. Multiple sclerosis in twins from continental Italy and Sardinia: a nationwide study. Ann. Neurol 2006; 59: 27-4.
- Islam T, Gauderman WJ, Cozen W, Mack TM. Childhood sun exposure influences risk of multiple sclerosis in monozygotic twins. Neurology. 2007; 69(4): 381-8.
- Robertson NP, O’Riordan JI, Chataway J, et al. Offspring recurrence rates and clinical characteristics of conjugal multiple sclerosis. Lancet 1997; 349: 1587–90.
- Ebers GC, Yee IML, Sadovnick AD, Duquette P and the Canadian Collaborative Study Group. Conjugal multiple sclerosis: population-based prevalence and recurrence risks in offspring. Ann Neurol 2000; 48: 927–31.
- Maghzi AH, Etemadifar M, Shaygannejad V, Saadatnia M, Salehi M, Hassanzadeh A. Conjugal multiple sclerosis in Isfahan, Iran: a population-based study. Mult Scler 2007; 13(5): 673-5
- Hawkes CH. Is multiple sclerosis a sexually transmitted infection? J Neurol Neurosurg Psychiatry 2002; 73(4): 439-43.
- Fernando M, Stevens CR, Walsh EC. Defining the role of the MHC in autoimmunity: A review and pooled analysis. PLOS genetics 2008; 4 (4): e1000024.
- Lincoln MR, Montpetit A, Cader MZ, Saarela J, Dyment DA, Tiislar M, Ferretti V, Tienari PJ, Sadovnick AD, Peltonen L et al.: A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet 2005, 37: 1108-12.
- Brum DG, Barreira AA, Louzada-Junior P, Mendes-Junior CT, Donadi EA. Association of the HLA-DRB1*15 allele group and the DRB1*1501 and DRB1*1503 alleles with multiple sclerosis in White and Mulatto samples from Brazil. J Neuroimmunol 2007 Aug 1 (En imprenta).
- Smestad C, Brynedal B, Jonasdottir G, Lorentzen AR, Masterman T, Akesson E, Spurkland A, Lie BA, Palmgren J, Celius EG, Hillert J, Harbo HF. The impact of HLA-A and -DRB1 on age at onset, disease course and severity in Scandinavian multiple sclerosis patients. Eur J Neurol 2007; 14: 835-40.
- Traherne JA, Barcellos LF, Sawcer SJ, Compston A, Ramsay PP, Hauser SL, Oksenberg JR, Trowsdale J. Association of the truncating splice site mutation in BTNL2 with multiple sclerosis is secondary to HLA-DRB1*15. Hum Mol Genet. 2006; 15: 155-61.
- Imrell K, Landtblom AM, Hillert J, et al. Multiple sclerosis with and without CSF bands: Clinically indistinguishable but immunogenetically distinct. Neurology 2006; 67: 1062-4.
- Dyment DA, Herrera BM, Cader MZ, Willer CJ, Lincoln MR, Sadovnick AD, Risch N, Ebers GC. Complex interactions among © Bajo licencia de Creative Commons Attribution 3.0 License Artículo disponible en: https://www.archivosdemedicina.com
- MHC haplotypes in multiple sclerosis: susceptibility and resistance. Hum Mol Genet. 2005; 14: 2019-26.
- Barcellos LF, Sawcer S, Ramsay PP, Baranzini SE, Thomson G, Briggs F, Cree BC, Begovich AB, Villoslada P, Montalban X, Uccelli A, Savettieri G, Lincoln RR, DeLoa C, Haines JL, Pericak-Vance MA, Compston A, Hauser SL, Oksenberg JR. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet. 2006; 15: 2813-24.
- Madsen LS, Andersson EC, Jansson L, Krogsgaard M, Andersen CB, Engberg J, Strominger JL, Svejgaard A, Hjorth JP, Holmdahl R et al.: A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet 1999; 23: 343-7.
- Khare M, Mangalam A, Rodriguez M, David CS: HLA DR and DQ interaction in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in HLA class II transgenic mice. J Neuroimmunol 2005; 169: 1-12.
- D. F. Uría. HLA y esclerosis múltiple. Estudios en la población española. Rev Neurol 2000; 31 (11): 1066-70.
- J. M. Zabay-Becerril, J. Burcet Dardé, J. Mulet-Ferrer, J. Soler Farré, C. Viader-Farré. Relación entre el alelo HLA-DRB1*1501 y la gravedad de la esclerosis múltiple en nuestra población española perteneciente a las Islas Baleares: influencia del sexo. Rev Neurol 2004; 38 (2): 118-22.
- De la Concha EG, Arroyo R, Crusius JB A, Campillo J A, Martin C, Valera de Sejias E et al. Combined effect of HLA-DRB1*1501 and interleukin-1 receotir antagoinist gene alele 2 in susceptibility to relapsing/remitting multiple sclerosis. J Neuroinmmunology; 80: 172-8.
- Olsson T, Hillert J. The genetics of multiple sclerosis and its experimental models. Curr Opin Neurol. 2008 Jun; 21(3): 255-60.
- International Multiple Sclerosis Genetics Consortium (IMSGC). Refining genetic associations in multiple sclerosis. Lancet Neurol 2008; 7 (7): 567-9.
- The International Multiple Sclerosis Genetics Consortium. Risk Alleles for Multiple Sclerosis Identified by a Genomewide Study. N Engl J Med 2007; 357: 851-62.
- Seboun E, Robinson MA, Doolittle TH, Ciulla TA, Kindt TJ, Hauser SL. A susceptibility locus for multiple sclerosis is linked to the T cell receptor beta chain complex. Cell 1989; 57: 1095–100.
- Hockertz M, Paty D, Beall S. Susceptibility to relapsing-progressive multiple sclerosis is associated with inheritance of genes linked to the variable region of the TCR _ locus: use of the affected familybased controls. Am J Hum Genet 1998; 62: 373–85
- Buhler MM, Bennets BH, Heard RNS, Stewart GJ. T cell receptor beta chain genotyping in Australian relapsing-remitting multiple sclerosis patients. Mult Scler 2000; 6: 140–7.
- Tienari PJ, Wikstrom J, Sajantila A, Palo J, Peltonen L. Genetic susceptibility to multiple sclerosis linked to myelin basic protein gene. Lancet 1992; 340: 987-91.
- Eoli M, Pandolfo M, Milanese C, Gasparini P, Salmaggi A, Zeviani M. The myelin basic protein gene is not a major susceptibility locus for multiple sclerosis in Italian patients. J. Neurol 1994; 241: 615-9.
- Eoli M, Wood NW, Kellar-Wood HF, Holmans P, Clayton D, Compston DAS. No linkage between multiple sclerosis and the T cell receptor alpha chain locus. J Neurol Sci 1994; 124: 32-7.
- Mycko MP, Kwinkowski M, Tronczynska E, Szymanska B, Selmaj KW. Multiple sclerosis: the increased frequency of the ICAM-1 exon 6 gene point mutation genetic type K469. Ann Neurol 1998; 44: 70-5.
- Vandenbroeck K, Opdenakker G, Goris A, Murru R, Billiau A, Marrosu MG. Interferon-gamma gene polymorphism--associated risk for multiple sclerosis in Sardinia. Ann Neurol 1998; 44: 841-2.
- Schrijver HM, Crusius JBA, Uitdehaag BMJ, Garcia Gonzalez MA, Kostense PJ, Polman CH, Pena AS. Association of interleukin-1-beta and interleukin-1 receptor antagonist genes with disease severity in MS. Neurology 1999; 52: 595-9.
- Jacobsen M, Schweer D, Ziegler A, Gaber R, Schock S, Schwinzer R, Wonigeit K, Lindert RB, Kantarci O, Schaefer-Klein J, Schipper HI, Oertel WH, Heidenreich F, Weinshenker BG, Sommer N, Hemmer B. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nature Genet 2000; 26: 495-9.
- Fazekas F, Strasser-Fuchs S, Kollegger H, Berger T, Kristoferitsch W, Schmidt H, Enzinger C, Schiefermeier M, Schwarz C, Kornek B, Reindl M, Huber K, Grass R, Wimmer G, Vass K, Pfeiffer KH, Hartung HP, Schmidt R. Apolipoprotein E epsilon4 is associated with rapid progression of multiple sclerosis. Neurology 2001; 57: 853-7.
- Chapman J, Vinokurov S, Achiron A, Karussis DM, Mitosek-Szewczyk K., Birnbaum M, Michaelson DM, Korczyn AD. APOE genotype is a major predictor of long-term progression of disability in MS. Neurology 2001; 56: 312-6.
- Mattila KM, Luomala M, Lehtimaki T, Laippala P, Koivula T, Elovaara I. Interaction between ESR1 and HLA-DR2 may contribute to the development of MS in women. Neurology 2001; 56: 1246-7.
- Garren H, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol 2008 May; 63(5):611-20.

