Keywords
Pompe; Lysosomal storage disorder; Glycogen; Exocytosis; Substrate degradation
Background
Pompe disease is caused by a deficiency in the lysosomal enzyme α-glucosidase (GAA) and results in the progressive accumulation of glycogen in autophagosome-autolysosome vesicles [1] and multivesicular bodies [2] in affected cells. There is currently no effective cure for this lysosomal storage disorder, but enzyme replacement therapy (ERT) has been clinically approved. ERT has been shown to reduce the amount of glycogen in a number of affected tissues, including heart and skeletal muscle [3], and there is evidence of prolonged patient survival [4]. However, it has been reported to be ineffective in some patients [3] and appears to have limited capacity to access the glycogen stored in some major sites of pathology, including type II skeletal muscle [1,5]. This inability of GAA to access the glycogen stored in certain autophagosomes-autolysosomes [6] necessitates the development of novel or adjunct therapeutic options to more effectively treat the disease.
The hallmark of Pompe disease is the accumulation of glycogen in autolysosomes, but a glucose tetrasaccharide (Glc4) has been observed in both the urine and blood of Pompe patients [7]. This product of glycogen breakdown is presumed to arise from digestion by circulating amylases [8] and to be the result of glycogen being released into circulation from affected cells. One possible explanation for this extracellular glycogen is cell death, and there is some evidence for apoptosis in the advanced stages of Pompe disease [9]. However, the amount of circulating tetrasaccharide in Pompe patients appears to be similar in both early- and late-stage disease [10], suggesting that cell death is at best only a partial explanation for this glycogen release from Pompe cells. An alternative explanation is the release of stored glycogen from muscle and other cells by exocytosis, a process that involves the fusion of intracellular vesicles with the cell surface and the release of vesicular content.
There is evidence that storage material can be released from lysosomal storage disorder cells by exocytosis. In kidney cells derived from metachromatic leukodystrophy patients, sulphatide storage material is released into the culture medium [11]. Exocytosis was induced in Niemenn Pick type C [12], multiple sulphatase deficiency, mucopolysaccharidosis (MPS) type-IIIA and neuronal ceroid lipofuscinoses cells [13], and resulted in a reduced amount of intracellular storage material. Recently, the induction of exocytosis in cultured Pompe cells was shown to cause a reduction in intracellular glycogen storage [13-15], suggesting that glycogen exocytosis is possible. Lysosomal exocytosis was induced by overexpressing transcription factor EB, which activated lysosomal Ca2+ channel MCOLN1 and increased the intracellular Ca2+ concentration. The modulation of exocytosis, by specific media and culturing conditions [16,17] as well as a number of drugs/compounds targeting the exocytic machinery [18,19], could also be utilized to modulate glycogen release. Here, a mass spectrometry based glycogen quantification assay [20] was used to accurately define glycogen exocytosis from Pompe skin fibroblasts under culture conditions that induce Ca2+-dependent exocytosis, confirming the work of Medina et al. [13], and Ca2+-independent exocytosis, identifying alternative targets with the potential to release a higher proportion of vesicular glycogen. We also defined the amount of exocytosis and glycogen released relative to other vesicular cargo and compared this process in fibroblasts from control, Pompe disease and MPS type I, a lysosomal storage disorder which accumulates different storage substrates. This led to the novel finding that although increased exocytosis in Pompe cells led to elevated glycogen release, it was partially impaired.
Materials and Methods
Cell culture
De-identified Pompe and MPS I skin fibroblasts were isolated from skin biopsies referred to the National Referral Laboratory for Lysosomal, Peroxisomal and Related Genetic Disorders (Women’s and Children’s Hospital, Adelaide, Australia) and used in accordance with Women’s and Children’s Human Research Ethics Committee approval 668/4/2009. Unaffected skin fibroblasts were derived from skin biopsies from apparently healthy volunteers. Each of the cultured fibroblast lines used had a similar rate of growth and was limited to less than nine subcultures.
Cytoplasmic glycogen depletion
To deplete the skin fibroblast cultures of cytoplasmic glycogen, cells were cultured in FBS- and glucose-free culture medium. For depletion, cells were then washed twice with 12 mL of PBS, pH 7.2 (Sigma, St. Louis, USA), and then cultured in FBS-free Basal Eagle medium (Sigma, St. Louis, USA) for 24 hours at 37°C. After discarding this culture medium, the cell monolayer was washed with 12 mL of PBS (three times) and cultured in glucose-free Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, USA) for 24 hours at 37°C.
Trypan blue cell viability measurement
To evaluate the viability of cultured cells at harvest, a 20 μL aliquot of cell suspension was mixed with an equal volume of 0.1% (v/v) trypan blue and incubated for 5 mins at 20°C, transferred to a haemocytometer and examined at 100X magnification. Nonviable cells were stained blue due to uptake of trypan blue into the cell. Culture viability was evaluated as the percentage of total cells that did not stain blue. Data were not collected from control fibroblast cultures with <90% trypan blue exclusion.
Lactate dehydrogenase cell viability measurement
A 300 μL aliquot of glucose-free DMEM from cultured cells was mixed with 200 μL of lactate dehydrogenase (LDH) assay substrate, 200 μL of LDH cofactor and 200 μL of LDH dye solution, and incubated for 30 mins at 20°C in the dark, in accordance with the TOX-7 kit instructions. Each reaction was stopped by the addition of 90 μL of 1 N HCl and analysed spectrophotometrically at both 690 nm (background signal) and 490 nm. A sample of glucose-free DMEM was included as a negative control. For the positive control, 10 mL of LDH assay lysis solution (diluted 1:10 in glucose-free DMEM) was added to a flask of cells to release cellular LDH. All assays, including the positive control, were performed in triplicate. The amount of LDH in the culture medium was corrected for total cell protein and expressed as the percentage of LDH released per culture. Data were not collected from control fibroblast cultures with >5 μg/mg of total cell protein LDH release.
Cell surface immune-fluorescence
Skin fibroblasts were seeded onto sterile coverslips in 6-well plates at approximately 1×104 cells/mL (each well containing 2 mL of complete culture medium). Cells were cultured to either 20% to 50% confluence (3.4 to 8.5×104 cells/well) or confluence` (1.7×105 cells/well). Each well was washed three times with 4 mL of PBS for 5 minutes at 4°C on a plate shaker. One hundred μL of mouse α-Lysosomal-associated membrane protein (LAMP)- 1 monoclonal antibody clone BB6 was then added to each well (diluted to 2.2 μg/mL in complete culture medium) and incubated for 1 hour at 4°C. Wells were then washed three times with 4 mL of PBS for 5 minutes at 4°C on a plate shaker. Each well was then aspirated and 100 μL of Fluorophore-488 conjugated goat α-mouse antibody (Invitrogen, Carlsbad, USA; diluted to 1:1000 in complete culture medium) was added and incubated in the dark for 1 hour at 4°C. All wells were washed three times with 3 mL of PBS for 5 minutes at 4°C on a plate shaker, in the dark. To stain the nucleus, 50 μL of Prolong Gold nuclear stain (Invitrogen, Carlsbad, USA), containing DAPI, was added to each coverslip. The coverslip was then inverted onto a microscope slide. Coverslips/ cells were stored in the dark at 4°C until examined on a Leica SP5 spectral scanning confocal microscope (Leica Microsystems Pty Ltd., North Ryde, Australia) at 100X magnification. Fluorescence intensity per unit area was determined using AnalySIS software (Soft Imaging System GmbH, Munster, Germany).
Intracellular immune-fluorescence
To visualise the intracellular location of LAMP-1 in skin fibroblasts, cells were permeabilised and stained by immune-fluorescence. To fix and permeabilise the cells, 1 mL of methanol/acetone (1:1) was added to each well (coverslip) and then incubated for 10 minutes at –20°C. Each well was then aspirated and air-dried for 20 minutes at 20°C. To prevent non-specific antibody binding, 1 mL of PBS containing 5% (w/v) bovine serum albumin (BSA; Sigma, St. Louis, USA) was added to each well and incubated for 1 hour at 20°C on a plate shaker. Each well was aspirated and 100 μL of monoclonal LAMP-1 antibody (diluted to 2.2 μg/mL in 5% (w/v) BSA in PBS) was added and incubated for 1 hour at 20°C. Each well was washed three times with 4 mL of PBS for 5 minutes at 4°C on a plate shaker. One hundred μL of Fluorophore-488 conjugated goat α-mouse secondary antibody (diluted to 1:200 in 5% (w/v) BSA in PBS) was added to each well and incubated in the dark for 1 hour at 20°C. All wells were washed three times with 4 mL of PBS for 5 minutes at 20°C on a plate shaker, in the dark. To stain the nucleus, 50 μL of Prolong Gold nuclear stain was added to each coverslip; the coverslip was then inverted onto a microscope slide. Coverslips/cells were stored in the dark at 20°C until examined on a Leica SP5 spectral scanning confocal microscope at 100X magnification.
Glycogen quantification
The mass spectrometry based glycogen assay has been described previously for the quantification of glycogen in mouse tissue extracts [20]. Here, the assay was applied to the quantification of glycogen in culture medium (100 μL analysed per sample) and skin fibroblast extracts (0.1 μg cell protein made up to 10 μL in water analysed per sample).
β-Hexosaminidase activity
The β-Hexosaminidase fluorometric assay was used as described previously [21].
Evaluation of cell division in skin fibroblasts
Approximately 1×106 cells were resuspended in 220 μL of propidium iodide solution, containing 0.5% (v/v) of Triton-X 100 (Sigma, St. Louis, USA), 250 μg/mL propidium iodide (Sigma, St. Louis, USA) and 250 μg/mL of RNase I (Sigma, St. Louis, USA) in PBS, and incubated in the dark for 30 minutes at 20°C. Different stages of the cell cycle were distinguished by the relative proportion of propidium iodide incorporated into the nucleus of each cell as described previously by Givan [22]. To remove excess propidium iodide solution, cells were washed with 1 mL of PBS for 1 minute on a plate shaker before final re-suspension in 1 mL of PBS. Cells were then injected into a FACScalibur flow cytometer (BD Biosciences, Franklin Lakes, USA) and the amount of propidium iodide intercalated into each cell was evaluated using CellQuest software (BD Biosciences, Franklin Lakes, USA); 20,000 cells were counted for each culture.
Statistical analysis
Differences between two independent groups of data with a normal distribution were determined by the student T-test, and the significance defined by a P value of <0.05. To compare three or more groups of data analysis of variance (ANOVA) was utilised, with P-values adjusted by the Holm's Stepdown Bonferroni procedure.
Results
Cell surface LAMP-1 staining of cultured skin fibroblasts
The integral lysosomal membrane protein LAMP-1 has previously been used to visualize vesicle exocytosis at the cell surface, by immune staining the luminal domain of this protein as it is exposed at the plasma membrane of live cells [23]. LAMP-1 fluorescence was observed as punctate staining on the cell surface of Pompe (Figure 1A), MPS I (Figure 1B) and unaffected cells (Figure 1C), with no apparent difference in fluorescence intensity and cell surface distribution among different cell types. Colchicine, a cytoskeletal destabiliser that inhibits exocytosis [23], caused a >70% reduction in cell surface LAMP- 1 fluorescence intensity on Pompe, MPS I and unaffected cells when compared to untreated cells (P <0.005; Figure 1D-F). There was no difference in cell surface LAMP-1 fluorescence intensity for Pompe, MPS I and unaffected cells treated with colchicine, while the absence of DAPI nuclear staining indicated that the cells had not been permeabilised. Permeabilised cells displayed an extensive network of larger LAMP-1-positive vesicular structures (Figure 1G-I), and DAPI nuclear staining.

Figure 1: Cell surface and intracellular LAMP-1 in cultured skin fibroblasts. Cell surface LAMP-1 staining was performed in non-permeabilised Pompe (A), MPS I (B) and unaffected (C) cells, and Pompe (D), MPS I (E) and unaffected (F) cells that had been pre-incubated in the presence of 1 nM of colchicine for 2 hours at 37°C. Intracellular LAMP-1 staining of permeabilised Pompe, MPS I and unaffected cells are shown in panels (G), (H) and (I), respectively. Images are an overlay of LAMP-1 fluorescence using the 488 channel (green) and DAPI (blue). Each image is representative of ≥ 20 images with each experiment performed in triplicate. Bar is equivalent to 5 μm.
β-Hexosaminidase exocytosis from cultured skin fibroblasts
The exocytosis of β-hexosaminidase has been used as a marker to track the release of the luminal content from vesicles fusing with the cell surface [24]. Fibroblast cells were cultured to confluence and either depleted or not depleted of cytoplasmic glycogen. The amount of intracellular β-hexosaminidase was 2-fold higher in Pompe (P <0.05) and 3.5-fold higher in MPS I when compared to unaffected cells, but there was no apparent difference in the amount of intracellular β-hexosaminidase in glycogen depleted cells, when compared to non-depleted cells (Figure 2). There was a rapid exocytosis of β-hexosaminidase from Pompe, MPS I and unaffected cells in the first 30 minutes of culture, which then plateaued over the next 1.5 hours (Figure 3A). Significantly more β-hexosaminidase was released from Pompe (2.2 ± 0.4 nmol/min/mg) and MPS I (3.8 ± 1.9 nmol/min/mg) cells than unaffected control cells (0.9 ± 0.4 nmol/min/mg; P <0.05). However, when the amount of β-hexosaminidase released was expressed as a percentage of total β-hexosaminidase in each cell type, Pompe, MPS I and unaffected cells released the same relative amount of this enzyme (approximately 2%; Figure 3B), suggesting β-hexosaminidase release was not impaired in Pompe and MPS I cells. Cell viability for each cell line was similar, when assessed by trypan blue exclusion ≥ 90% and LDH release <5 μg/ mg of total cell protein.

Figure 2: Intracellular amounts of β-hexosaminidase (β-hex) in cultured skin fibroblasts. The amount of β-hexosaminidase was determined in cell extracts derived from non-depleted (total) and depleted (vesicular) Pompe (□), MPS I (×) and unaffected (Δ) cells (cell lines were from different individual patients or control subjects). The activity of β-hexosaminidase is presented as nmol/min/mg of total cell protein. *Indicates cell lines used in Figure 5 onwards.

Figure 3: β-Hexosaminidase release from cultured skin fibroblasts. The release of β-hexosaminidase (β-hex) was determined from glycogen-depleted Pompe (red), MPS I (green) and unaffected (black) cells (cell lines were from different individual patients or control subjects; and (Δ)). All cells were cultured in glucose-free DMEM from t=0 to 2 hours. In panel (A), results are expressed as nmol/min/mg of β-hex activity released into the culture medium (mean ± standard deviation (n=3)). In panel (B) results are expressed as the percentage of total β-hex activity in the culture medium (mean ± standard deviation (n=3)). *Significant difference (P<0.05) when compared to unaffected cells.
Glycogen exocytosis from cultured skin fibroblasts
There were similar amounts of total cellular glycogen in nonglycogen- depleted Pompe, MPS I and unaffected cells (Figure 4). However, in the glycogen depleted cells (have mainly vesicular glycogen), Pompe cells contained 88 ± 5 μg/mg of glycogen, MPS I cells had 47 ± 11 μg/mg of glycogen and unaffected cells had 20 ± 5 μg/mg of glycogen (P <0.05 for all comparisons). The amount of glycogen released from glycogen depleted Pompe cells was 1.4-fold greater than glycogen depleted MPS I cells (P <0.05) and 2.7-fold greater than glycogen depleted unaffected control cells (P <0.005; Figure 5A). Although releasing significantly more glycogen, the percentage of glycogen released from Pompe cells was >30% less than that observed for MPS I (P <0.05) and unaffected control cells (P <0.005; Figure 5B).
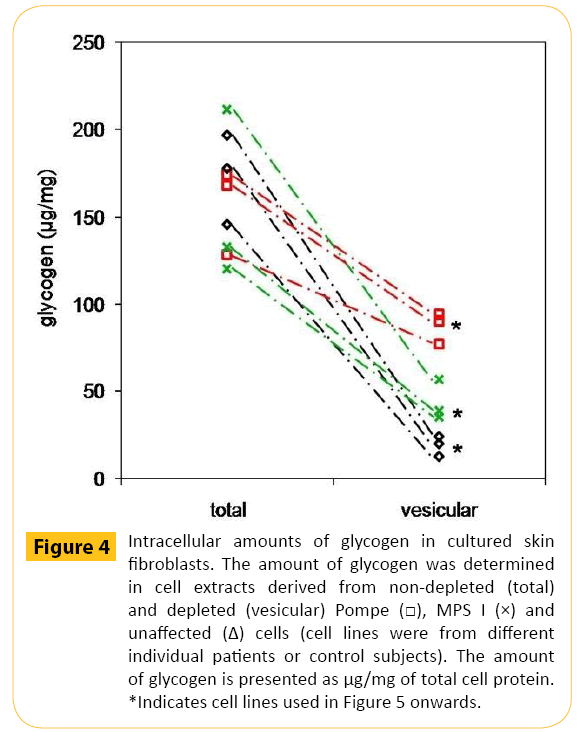
Figure 4: Intracellular amounts of glycogen in cultured skin fibroblasts. The amount of glycogen was determined in cell extracts derived from non-depleted (total) and depleted (vesicular) Pompe MPS I (×) and unaffected (Δ) cells (cell lines were from different individual patients or control subjects). The amount of glycogen is presented as μg/mg of total cell protein. *Indicates cell lines used in Figure 5 onwards.

Figure 5: Glycogen release from cultured skin fibroblasts. Glycogen release was determined from cytoplasmic glycogen-depleted Pompe (red), MPS I (green) and unaffected (black) cells (cell lines were from different individual patients or control subjects; and (Δ)). All cells were cultured in glucose-free DMEM from t=0 to 2 hours. In panel (A), results are expressed as μg/mg of glycogen released into the culture medium (mean ± standard deviation (n=3)). In panel (B), results are expressed as the percentage of total glycogen in the culture medium (mean ± standard deviation (n=3)). *Significant difference (P<0.05) when compared to unaffected cells. **Significant difference (P<0.005) when compared to unaffected cells. Panel (C) shows a comparison of the amount of vesicular glycogen released from cytoplasmic glycogen-depleted Pompe, MPS I and unaffected cells. Data are presented as μg of glycogen in the culture medium per mg of total cell protein or the total percentage of glycogen released into the culture medium.
Glycogen and β-hexosaminidase exocytosis from colchicine-treated skin fibroblasts
Colchicine-treated Pompe, MPS I and unaffected cells (glycogen depleted) showed a ≥ 20% reduction in the release of β-hexosaminidase and glycogen into the culture medium after two hours, when compared to untreated control cells (P <0.03; Figure 6). Trypan blue exclusion was ≥ 90% and LDH release <5 μg/mg of total cell protein for each treatment group.

Figure 6: Effect of colchicine on the release of β-hexosaminidase and glycogen from cultured skin fibroblasts. The release of β-hexosaminidase (β-hex) and glycogen was determined from cytoplasmic glycogen-depleted Pompe (A, B; red), MPS I (C, D; green) and unaffected (E, F; black) cells. Cells were treated with 1 nM of colchicine in glucose-free DMEM for 2 hours at 37°C (□) or untreated (■). Results are expressed as the percentage of β-hex/glycogen in the culture medium (mean ± standard deviation (n=3)). *Significance P<0.05 when compared to untreated controls. **Significance P<0.005 when compared to untreated controls.
The effect of Ca2+ on cell surface LAMP-1 staining, and β-hexosaminidase and glycogen exocytosis from Pompe skin fibroblasts
Approximately 2-fold more glycogen was released into the culture medium from Pompe fibroblast cells after treatment with 2.3 mM of extracellular Ca2+, when compared to 1.8 mM of extracellular Ca2+ (P <0.05; Figure 7A). The amount of β-hexosaminidase released into the culture medium of Pompe cells increased after treatment with 2.3 mM of extracellular Ca2+, but this was not significantly more when compared to 1.8 mM of extracellular Ca2+ (P >0.05; Figure 7B). Trypan blue exclusion was ≥ 90% and LDH release was <5 μg/mg of total cell protein for Ca2+ concentrations of both 1.8 mM and 2.3 mM. The amount of LDH release was more variable with 2.3 mM Ca2+ treatment (3.2 ± 2.5 μg/mg), as was trypan blue exclusion (93.8 ± 3.6% of total cells), when compared to 1.8 mM Ca2+ treatment (93.3 ± 1.3% of total cells). Pompe cells treated with 3.6 mM Ca2+ displayed increased cell permeability, suggesting some cell death, with LDH release at 10.1 ± 2.1 μg/mg and trypan blue exclusion at 79.2 ± 4.4% of cells.

Figure 7: The effect of Ca2+ on exocytosis in Pompe skin fibroblasts. Panel (A, B) shows the effect of extracellular Ca2+ on Pompe skin fibroblast exocytosis. Pompe cells were treated with glucose-free DMEM containing 1.8 mM and 2.3 mM of CaCl2. CaCl2 supplemented DMEM was added to the cells at t=0; cells were then incubated for 2 hours. The release of glycogen (panel A) and β-hexosaminidase (β-hex; panel B) into the culture medium was determined. Panel (C, D) shows the effect of intracellular Ca2+ on Pompe skin fibroblast exocytosis. Pompe cells were treated with 10 μM of BAPTA-AM. The BAPTA-AM-supplemented DMEM was added to the cells at t=0 and cells were incubated for 2 hours. The release of glycogen (panel C) and β-hexosaminidase (panel D) into the culture medium of BAPTA-AM-treated and untreated cells was determined. Results are expressed as the total percentage released into the culture medium (mean ± standard deviation (n=3)). *Significance P<0.05 and **significance P<0.005 compared to untreated control cells. Cell surface LAMP-1 fluorescence of non-permeabilised BAPTA-AM-treated and untreated cells is shown in panels (E) and (F), respectively. Images are an overlay of LAMP-1 fluorescence (green) and DAPI (blue). Each image is representative of >20 images. Size bar equivalent to 5 μm.
Pompe skin fibroblasts treated with the Ca2+ chelator, 1,2-Bis(2- aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakisacetoxymethyl ester (BAPTA-AM), which decreases the concentration of cytosolic Ca2+, showed a ≥ 65% reduction in the amount of glycogen (Figure 7C) and β-hexosaminidase (Figure 7D) released into the culture medium, when compared to untreated cells (P <0.05; at two hours). There was no significant difference in the amount of LDH released into the culture medium (3.1 ± 0.6 μg/mg) or trypan blue exclusion (93.8 ± 2.4% of total cells) in cells treated with BAPTA-AM, when compared to untreated cells. BAPTA-AM-treated cells demonstrated an 20% reduction in the fluorescence intensity of external plasma membrane LAMP-1 staining compared to untreated cells (Figure 7E, F; P <0.05), while DAPI nuclear staining was similar.
The effect of culture confluence on cell surface LAMP-1, β-hexosaminidase and glycogen exocytosis from Pompe skin fibroblasts
The amount of exocytosis and glycogen release was evaluated in Pompe cells at different stages of confluence. Cell cultures at pre-confluence contained a higher percentage of dividing cells, when compared to Pompe cells at confluence and the percentage of dividing cells remained at 5% for Pompe cells from 90% confluence through to three weeks post-confluence (Figure 8A). There was a >3.6-fold increase in the amount of glycogen released into the culture media in cells that had not yet reached confluence (40% and 70%), when compared to one week post-confluence (P <0.001 at two hours; Figure 8B). There was a greater than 6-fold increase in the amount of β-hexosaminidase released from cells at 40% confluence and 70% confluence, when compared to cells at one week post-confluence (P <0.001 at two hours; Figure 8C). Cell viability was measured at each stage of confluence with trypan blue exclusion ≥ 90% and LDH release <5 μg/mg of total cell protein.

Figure 8: The effect of Pompe skin fibroblast culture confluence on exocytosis. The percentage of Pompe cells at each stage of the cell cycle was determined for cells from 20% confluence to three weeks post-confluence (A). Nonglycogen- depleted cells were cultured in DMEM (10% FBS) until harvested. The percentage of Pompe cells in the G0/G1 phase ; senescent), S phase (◊; growth phase) and G2/M phase (Δ; growth phase) is shown. Data for stages S and G2/M were also combined to provide a total percentage of cells in the growth phase . Each data point was evaluated from the measure of 20,000 Pompe cells by flow cytometry. The release of glycogen (B) and β-hexosaminidase (β-hex; C) was evaluated in non-glycogen-depleted Pompe cells at 40% confluence , 70% confluence and one week post-confluence (◊). Glucose-free DMEM was added to the cells at t=0 and cells were incubated for 2 hours. Results are expressed as the mean ± standard deviation (n=3). **Significance P<0.005 when compared to cells at confluence.
Pompe, MPS I and unaffected cells at 40% confluence displayed punctate cell surface LAMP-1 staining that was localised to distinct areas of each cell (Figure 9A-C); which was associated with ruffling and filopodia (also known as microspikes; Figure 9J and Figure 9K for Pompe cells). The amount of cell surface LAMP-1 staining in Pompe, MPS I and unaffected cells at 40% confluence was 1.6- fold higher, when compared to cells at confluence (P <0.05; Figure 9L for Pompe cells). Pompe, MPS I and unaffected cells at 40% confluence were permeabilised by fixation and displayed a more extensive staining pattern and larger vesicular structures than nonpermeabilised cells (Figure 9G-I). Pompe, MPS I and unaffected cells at 40% confluence were also treated with colchicine, which showed a >30% reduction in the fluorescence intensity of cell surface LAMP-1 punctae in colchicine-treated (10 minutes), when compared to untreated cells (P <0.05; Figure 9D-F). The release of β-hexosaminidase and glycogen from colchicine treated cells at 40% confluence could not be evaluated for ≥ 30 minutes because of a significant reduction in cell viability (≥ 6.9 μg/mg LDH and ≤ 82.3% trypan blue exclusion). In non-permeabilised cells treated with colchicine for <30 minutes, trypan blue exclusion was ≥ 90% and LDH release was <5 μg/mg of total cell protein.

Figure 9: The effect of confluence on cell surface LAMP-1 staining. LAMP-1 staining was evaluated in non-permeabilised (cell surface) and permeabilised (intracellular) Pompe, MPS I and unaffected cells at 40% confluence. Cells were non-glycogen-depleted and cultured in DMEM (10% FBS) until harvested. Cell surface LAMP-1 staining was evaluated in Pompe (A, J, K), MPS I (B) and unaffected (C) cells at 40% confluence. Cell surface LAMP- 1 staining was also measured in colchicine-treated (1 nM) Pompe (D), MPS I (E) and unaffected (F) cells at 40% confluence (10 min incubation only). Intracellular LAMP-1 fluorescence was determined in permeabilised Pompe (G), MPS I (H) and unaffected (I) cells at 40% confluence. Cell surface LAMP-1 staining of Pompe (l) skin fibroblasts at confluence (i.e. cell-to-cell contact) is also presented. Images are an overlay of LAMP- 1 fluorescence (green), DAPI (blue) and DIC. Each picture represents >20 images with each experiment performed in triplicate. Size bar equivalent to 10 μm.
Discussion
Exocytosis is not impaired in Pompe and MPS I fibroblasts
Exocytosis involves the fusion of vesicles with the cell surface to deliver luminal cargo to the extracellular milieu. LAMP-1- positive punctae have been used to define Ca2+-triggered exocytic events at the cell surface of non-permeabilised NRK cells, keratinocytes and fibroblasts [13,25]. Newly synthesised LAMP- 1 is transported from the trans-Golgi network to endosomes/ lysosomes via intracellular vesicles [26]. Surface expression of LAMP-1 occurs when endosomal/lysosomal vesicles fuse with the plasma membrane exposing the luminal face of the vesicle [26]. The fluorescence intensity and cell surface distribution of LAMP-1 was similar in Pompe, MPS I and unaffected skin fibroblasts, suggesting that there were similar amounts of these exocytic events in these different cell types. The activity of β-hexosaminidase in cell extracts and culture medium has been used to determine the amount of exocytosis from cultured cells [27]. β-Hexosaminidase can be released from the cell through the constitutive secretory pathway [28], however, for short term studies, based on the biosynthetic rate of β-hexosaminidase, the amount of enzyme release via this pathway is expected to be minimal (≤ 3% after 24 hours in culture) [29]. While the amount of intracellular β-hexosaminidase varied between Pompe, MPS I and unaffected cells the relative percentage of β-hexosaminidase released into the culture medium after two hours was similar for all three cell lines (approximately 2%) and was consistent with previous reports for unaffected cultured fibroblasts (1.5 to 5%) [15,30]. The amount of exocytosis from Pompe and MPS I cells was therefore similar to unaffected control cells suggesting that the two lysosomal storage disorder cell types did not have any impairment in the process of exocytosis. In addition, Pompe, MPS I and unaffected control cells all released glycogen into the culture medium, which was consistent with previous reports of Ca2+- dependent exocytosis and the release of lysosomal content [27].
Glycogen is effectively exocytosed from Pompe fibroblasts
The treatment of cultured cells with colchicine, a cytoskeletal destabiliser, has been reported to inhibit exocytosis through its action on microtubules and this is thought to impede the traffic of vesicles to the cell surface [24]. Colchicine treatment of Pompe, MPS I and unaffected cells led to a >30% reduction in the exposure of LAMP-1 at the cell surface, and also the release of β-hexosaminidase and glycogen, but with no change in cell viability. The release of β-hexosaminidase was similar to that observed in NRK cells, with colchicine treatment leading to a 15% reduction in the exocytosis of this enzyme [30]. Treatment of fibroblast cells with colchicine was able to reduce the amount of glycogen released from Pompe, MPS I and unaffected control cells supporting the notion that this was being released by exocytosis. In cultured Pompe, MPS I and unaffected fibroblast cells, there was a rapid increase in the amount of β-hexosaminidase released into the culture medium for the first 30 minutes, followed by a plateau in the release. For exocytosis from NRK cells [24], fibroblasts [12] and mast cells [14] there was also a plateau in the amount of β-hexosaminidase released after time in culture. Extracellular β-hexosaminidase can be re-internalised into fibroblasts by endocytosis [29] and may therefore contribute to this apparent plateauing effect. Importantly, glycogen release did not plateau to the same extent as β-hexosaminidase release and this may indicate that there is not a specific uptake mechanism for glycogen in fibroblasts.
Ca2+ concentration affects glycogen exocytosis in Pompe fibroblasts
The concentration of Ca2+ in the culture medium had a significant impact on the amount of glycogen exocytosis from Pompe cells. Increasing the concentration of Ca2+ in the culture medium has been reported to facilitate the release of acid hydrolases from cells [15], whereas decreasing the concentration of Ca2+ reduces this exocytic release [31]. There was a 70% increase in the release of β-hexosaminidase from Pompe cells in response to a higher concentration of Ca2+; but a 300% increase in β-hexosaminidase release has been reported in NRK cells [15]. Similarly, there was a 65% reduction in the release of β-hexosaminidase from Pompe cells in response to BAPTA-AM, whereas NRK cells displayed a >90% reduction [32]. Cultured fibroblasts may therefore be less responsive to Ca2+ than other cell types. Despite these differences in β-hexosaminidase release, modulation of Ca2+-dependent exocytosis was able to alter glycogen exocytosis, confirming the work of Medina et al. [13].
Cell confluence affects glycogen exocytosis in Pompe fibroblasts
Pompe cells undergoing cell division released >75% of the total cell β-hexosaminidase and >80% of the glycogen into the culture medium after only 2 hours of culture, which was >7-fold higher than that observed for confluent cells. This high amount of glycogen release was surprising as Pompe cells only contain approximately 50% vesicular glycogen (single point in time measurement), with the remainder being localised to the cytoplasm. A potential explanation for this high percentage of glycogen release may be that during cell growth cytoplasmic glycogen is rapidly autophagocytosed to provide an energy source to drive these processes, thereby depleting the cytoplasmic stock of glycogen and providing a pool of vesicular glycogen that is susceptible to exocytic release. Increased glycogen autophagy provides an energy source in liver cells during the high energy demand post-natal period [33].
Up-regulation of glycogen exocytosis in subconfluent Pompe fibroblasts correlates with increased cell migration
Cell division and migration are elevated in cultures at preconfluence [34]. Increased exocytosis has been reported during cell division and occurs at the cleavage furrow, as cells divide [35]. Exocytosis also occurs during cell migration and this involves membrane localised to the leading edge of the cell [36]. There was no obvious increase in cell surface LAMP-1 staining at the cleavage furrow of dividing Pompe cells. Golgi-derived vesicles, but not endosomal or lysosomal compartments, have been linked to increased exocytosis during cell division [35], implicating Ca2+-independent exocytosis. In sub-confluent, migrating Pompe cells, there was an increase in cell surface LAMP-1 staining at the leading edge of the cell, in areas of ruffling and on filopodia. These structures are involved in the transfer of membrane from an intracellular location to the cell surface, thereby enabling forward motion [36]. The elevated exocytosis associated with sub-confluent Pompe skin fibroblasts, as measured by increased LAMP-1 staining and β-hexosaminidase release, may therefore be related to cell migration rather than cell division per se.
Glycogen release from Pompe and MPS I fibroblasts is partially impaired
When expressed as a percentage of total cellular glycogen for each cell, Pompe cells released the lowest proportion of vesicular glycogen compared to MPS I and unaffected cells (Figure 5C). However, the amount of exocytosis for each of these cell lines was similar, as determined by the presence of cell surface LAMP- 1 staining and the release of β-hexosaminidase into the culture medium. This suggested that the reduced glycogen release from Pompe (and to a lesser extent MPS I) cells was not due to impaired exocytosis per se. One possible explanation is that Ca2+- induced exocytosis, which is the likely mechanism responsible for glycogen release, results in cavicapture [37], the partial release of vesicle content through an exocytic pore. An explanation for this reduced glycogen exocytosis may be that the glycogen granules in Pompe cells may be larger in diameter than those in unaffected cells, and therefore limited in their ability to be exocytosed. Alternatively, the diameter of the exocytic pore in Pompe cells may be restricted, when compared to unaffected cells, which would also limit glycogen release. These two possible mechanisms for the reduced release of glycogen from Pompe cells are the focus of future studies.
MPS I fibroblasts accumulate glycogen
MPS I cells were included in this study as these cells accumulate different storage substrates to Pompe cells, namely glycosaminoglycans. However, MPS I cells were shown to contain more glycogen than unaffected cells, indicating elevated vesicular glycogen stores. While the defect in GAA explains increased glycogen in Pompe cells, a defect in α-L-iduronidase would not be expected to result in glycogen storage. It remains unknown as to why glycogen accumulates in MPS I cells, and further studies are required. However, it could be speculated that glycosaminoglycan storage in MPS I cells either impairs the catalytic activity in endosome-lysosome compartments or limits lysosomal fusion and therefore degradation in autolysosomes
Conclusion
In this study, we showed no impairment of exocytosis in Pompe cells as defined by cell surface LAMP-1 staining and β-hexosaminidase release; and that glycogen was exocytosed from these cells. Importantly, glycogen release from Pompe fibroblasts was up-regulated by certain culture conditions, presumably due to specific effects on the exocytic machinery. This study provided proof of concept that glycogen exocytosis may be used to re-locate storage material from Pompe cells into the extracellular milieu. The induction of Ca2+-independent exocytosis appeared to release more glycogen from Pompe cells than Ca2+- dependent exocytosis. A greater understanding of the Ca2+- independent exocytic mechanism may lead to the identification of new therapeutic targets, which may be more beneficial than those identified for the induction of Ca2+-dependent exocytosis [13]. The induction of glycogen exocytosis opens the possibility for an alternative therapeutic strategy based on degradation by circulating amylases.
Acknowledgements
The authors gratefully acknowledge funding from the Masonic Foundation. C. Turner was funded by the NHMRC Dora Lush Scholarship.
Funding
The authors gratefully acknowledge funding from the Masonic Foundation. C. Turner was funded by the NHMRC Dora Lush Scholarship.
6759
References
- Qureshi OS, Paramasivam A, Yu JC, Murrell-Lagnado RD (2007) Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J Cell Sci120:3838-3849.
- Cardone M, Porto C, Tarallo A, Vicinanza M, Rossi B, et al. (2008) Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics1(1):6.
- Kishnani PS, Beckemeyer AA (2014) New therapeutic approaches for Pompe disease: enzyme replacement therapy and beyond. PediatrEndocrinol Rev. 12 Suppl 1:114-124.
- Sugo T, Tachimoto H, Chikatsu T, Murakami Y, Kikukawa Y, et al. (2006) Identification of a lysophosphatidylserine receptor on mast cells. BiochemBiophys Res Commun341:1078-1087.
- Hawes ML, Kennedy W, O'Callaghan MW, Thurberg BL (2007) Differential muscular glycogen clearance after enzyme replacement therapy in a mouse model of Pompe disease. Mol Genet Metab91:343-351.
- Sesaki H, Ogihara S (1997) Protrusion of cell surface coupled with single exocytotic events of secretion of the slime in Physarum plasmodia. J Cell Sci 110:809-818.
- Rodríguez A, Martinez I, Chung A, Berlot CH, Andrews NW (1999) cAMP regulates Ca2+-dependent exocytosis of lysosomes and lysosome-mediated cell invasion by trypanosomes. J BiolChem274:16754-16759.
- Kumlien J, Andrén-Sandberg A, Zopf D, Lundblad A (1989) Determination of a glucose-containing tetrasaccharide in urine of patients with acute pancreatitis. Int J Pancreatol 4:139-147.
- Hesselink RP, Wagenmakers AJ, Drost MR, Van der Vusse GJ (2003) Lysosomal dysfunction in muscle with special reference to glycogen storage disease type II. BiochimBiophysActa1637:164-170.
- An Y, Young SP, Kishnani PS, Millington DS, Amalfitano A, et al. (2005) Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab85:247-254.
- Klein D, Büssow H, Fewou SN, Gieselmann V (2005) Exocytosis of storage material in a lysosomal disorder. BiochemBiophys Res Commun 327:663-667.
- Chen FW, Li C, Ioannou YA (2010) Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One. 5:e15054.
- Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, et al. (2000) Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. 148:1141-1149.
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA et al. (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 5:691-706.
- Martina JA, Diab H, Lishu L, Jeong-A L, Patange S, et al. (2014) The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 21:7.
- Barg S, Macado JD (2008) Compensatory endocytosis in chromaffin cells. Acta Physiol 192:195-201.
- Rodríguez A, Webster P, Ortego J, Andrews NW (1997) Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137:93-104.
- Jaiswal JK, Andrews NW, Simon SM (2002) Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol 159:625-635.
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, et al. (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell21:421-430.
- Fuller M, Duplock S, Turner C, Davey P, Brooks DA, et al. (2012) Mass spectrometric quantification of glycogen to assess primary substrate accumulation in the Pompe mouse. Anal Biochem. 421:759-763.
- Leaback DH, Walker PG (1961) Studies on glucosaminidase. 4. The fluorimetric assay of N-acetyl-beta-glucosaminidase. Biochem J 78:151-156.
- Givan AL (2001) Flow cytometry: first principles 2ndedn. Wiley, John & Sons, Inc. New York.
- Pan CY, Lee H, Chen CL (2006) Lysophospholipids elevate [Ca2+]i and trigger exocytosis in bovine chromaffin cells. Neuropharmacology. 51:18-26.
- Kuncl RW, Bilak MM, CraigSW, Adams R (2003) Exocytotic "constipation" is a mechanism of tubulin/lysosomal interaction in colchicine myopathy. Exp Cell Res 285:196-207.
- LaPlante JM, Sun M, Falardeau J, Dai D, Brown EM, et al. (2006) Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab. 89:339-348.
- Eskelinen EL, Tanaka Y, Saftig P (2003) At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol13:137-145.
- Shea L Raben N (2009) Autophagy in skeletal muscle: implications for Pompe disease. Int J ClinPharmacolTher. 47 Suppl 1:S42-S47.
- Rozaklis T, Ramsay SL, Whitfield PD, Ranieri E, Hopwood JJ, et al. (2002) Determination of Oligosaccharides in Pompe Disease by Electrospray Ionization Tandem Mass Spectrometry. ClinChem48:131-139.
- Tiwari N, Wang CC, Brochetta C, Ke G, Vita F, et al. (2008) VAMP-8 segregates mast cell-preformed mediator exocytosis from cytokine trafficking pathways. Blood 111:3665-3674.
- Raben N, Fukuda T, Gilbert AL, de Jong D, Thurberg BL, et al. (2005) Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. MolTher 11:48-56.
- Li D, Ropert N, Koulakoff A, Giaume C, Oheim M (2008) Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J Neurosci 28:7648-7658.
- Ito N, Yokomizo T, Sasaki T, Kurosu H, Penninger J, et al. (2002) Requirement of phosphatidylinositol 3-kinase activation and calcium influx for leukotriene B4-induced enzyme release. J BiolChem 277:44898-44904.
- Kotoulas OB, Kalamidas SA, Kondomerkos DJ (2004) Glycogen autophagy. Microsc Res Tech 64:10-20.
- Boucrot E, Kirchhausen T (2007) Endosomal recycling controls plasma membrane area during mitosis. Proc NatlAcadSci USA 104:7939-7944.
- Goss JW, Toomre DK (2008) Both daughter cells traffic and exocytose membrane at the cleavage furrow during mammalian cytokinesis. J Cell Biol 181:1047-1054.
- Sagherian C, Thorner P, Mahuran D (1994) The pro-peptide of the pro beta-polypeptide chain of human beta-hexosaminidase is necessary for proper protein folding and exit from the endoplasmic reticulum. BiochemBiophys Res Commu. 204:135-141.
- Jaiswal JK, Chakrabarti S, Andrews NW, Simon SM (2004) Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoSBiol 2:233.

