Iezzi ML*, Varriale G, Zagaroli L, Greco M, Lasorella S and Verrotti di Pianella A
Pediatric Department, University of L’Aquila, San Salvatore Hospital, Italy
*Corresponding Author:
Maria Laura Iezzi
Pediatric Department, University of L’Aquila
San Salvatore Hospital, Italy
Tel: 00390862368207
Email: marialaura.iezzi@libero.it
Received Date: 20 February 2017 Accepted Date: 25 March 2017 Published Date: 30 March 2017
Citation: Iezzi ML, Varriale G, Zagaroli L, et al. Growth Hormone Deficit as a First Clinical Manifestation of Early Diffuse Cerebral Glioma in Patient with Neurofibromatosis. Ann Clin Lab Res. 2017, 5: 1.
Introduction
Neurofibromatosis type 1 (NF1) is a new term that entirely replaces the old name von Recklinghausen’s disease. It is the most common among the forms of neurofibromatosis (NF). NF1 has an estimated incidence of 1:2500 live births and a prevalence of 1:4000 in the general population. It is an autosomal dominant disease, showing an intrafamilial phenotype expressivity. Clinically, NF1 is characterized by the presence of multiple (>6) cafè-au-lait spots and at least one of the following signs: axillary freckling (Crowe’s sign), Lisch nodules, cutaneous or subcutaneous neurofibromas, carried by first-degree relatives affected by this disease. Other clinical signs, defined as minor criteria, are the following, in decreasing order: macrocefalia, growth failure, thoracic malformations, cortical thinning of long bones, small angiomas (Campbell de Morgan spots) located in the thorax or thighs. NF1 may exhibit a wide range of complications; particularly neurological, such as, symptomatic optic pathway tumors, cerebral gliomas, symptomatic cerebral acqueductal stenosis and spinal compression due to intraspinal NF. The glioma is relatively a benign tumor occurring in approximately 15% of the carriers of neurofibromatosis, frequently expressed in the offspring of carriers, showing an onset between 4-6 years of age. The development of glioma is generally slow and asymptomatic. In 1% to 5% of cases it becomes symptomatic, especially in sporadic forms of NF, shown by deficient visual acuity, strabismus, pupillary and other cerebral abnormalities. Secondary to pituitary damage, the progression of the mass will sometimes occur with endocrinological signs such as precocious or delayed puberty, tertiary hypothyroidism and GH deficiency (GHD) [1].
We need to point out that GHD, included as a minor criterion for the NF, does not correlate with disease severity and is multifactorial, stemming from the disease itself or its complications such as suprasellar lesions, surgery or radiotherapy for intracranial lesions.
Patient Presentation
A 2 years old girl came to our pediatric endocrinology center for delay of growth and anorexia. Her mother, who showed 4 cafè-au-lait spots, was previously surgically treated for pituitary adenoma. On examination, the child showed: stature <1° centile (C), according to growth curves of Tanner, body mass index (BMI) 14 (5° C), presence of 10 café-au-lait spots spread over the whole body, altered positioning of the toes of the left foot, squint right eye, and epicanthic folds.
Diagnostic Investigations
Hemoglobin (Hb): 11.7 gr/dl, ferritin: 3.98 ngr/ml, Insulin like growth factor-1 (IGF1): 110 ngr/ml, Thyroid Stimulating Hormone (TSH): 0.35 mcU/ml, Free Thiroxin 4 (fT4) 1.08 mcU/ml, GH peak after stimulus with clonidine: 7 ngr/ml, as shown in Table 1. The remainder of her laboratory examinations were unremarkable. Genetic tests for NF in both the child and mother were suggested.
| Laboratory Findings |
Results |
Normal Laboratory References |
| Hb |
11.8 gr/dl |
12.5 gr/dl to 14.5 gr/dl |
| Ferritin |
3.98 ngr/ml |
20 ngr/ml to 280 ngr/ml |
| TSH |
0.35 mcU/ml |
0.35 mcU/ml to 4.94 mcU/ml |
| fT4 |
1.08 mcU/ml |
0.7 mcU/ml to 1.48 mcU/ml |
| IGF1 |
110 ngr/ml |
247 ngr/ml to 481 ngr/ml |
| GH peak |
7 ngr/ml |
<8 ng/ml |
Table 1: Laboratory results vs normal laboratory references.
While waiting for a planned magnetic resonance image (MRI) brain scan, a second test for diagnosing GHD and a check of thyroid function were made. Results confirmed GHD and a reduction in TSH secretion with a preserved thyroid function. In the meantime, the child showed worsening signs of anorexia and weight loss and several crises of hypotonia with falling to the ground. Upon neurological examination, the child showed psychomotor delay, ataxia, nystagmus, symmetrical hypotonia with hyperreflexia, in addition to the cafè-au-lait spots, compatible with NF1.
An urgent MRI brain scan showed diffuse and bihemispheric leukoencephalopathy particularly evident in the pontomesencefalic, the temporal and frontal-parietal periventricular areas. Altered signals and morphology of both of the optic nerves with a stronger prevalence in the chiasma and optic radiations. No evidences of atrophy of pituitary gland. The described findings are compatible with "gliomatosis cerebri" (Figure 1). The child was immediately transferred to the department of Pediatric Oncology, in which the glioma was treated with radiotherapy and then chemotherapy. She is still living and continues treatment, but we haven’t precise informations of her neurological and clinical follow-up.Discussion
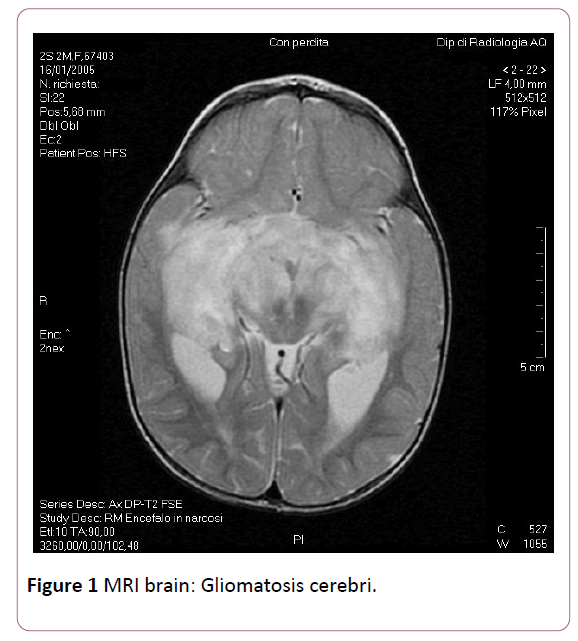
Figure 1: MRI brain: Gliomatosis cerebri.
Discussion
One of the peculiarities of this case report is the particular aggressiveness of the glioma and its early onset. It is important to stress that MRI is a basic step in the diagnostic pathway of GHD because it can detect organic causes of disease, in particular, the presence of intracranial tumors, as in our case report. In 99% of the familiar forms of NF1 the glioma progresses very slowly and therefore is asymptomatic for a long time; besides, as already specified, the classical occurrence of this complication is in children between 4 and 6 years of age [2,3].
Moreover, the child’s arrested growth was the clinical sign that led the parents to consult a specialized center and this triggered a series of diagnostic tests based on the visit and the family’s history. The studies in literature suggest that a GHD screening is necessary in children affected by NF1 and short stature [4,5] GHD is an important complication in children with NF1, with the etiology in some patients remaining unclear. In the majority of children with NF1, GHD occurs primarily in those with an intracranial tumor who undergo intracranial surgery and cranial irradiation therapy.
However, Cnossen et al. reported a 2.5% prevalence of GHD in children with NF1 without an intracranial mass and before surgical or irradiation therapy for optic pathway gliomas (OPG), a frequency that is significantly higher than the 0.03% observed in the general pediatric population [6]. An OPG was detected in 1 of 3 children with GHD suggesting that GHD appears independently of the presence of OPG. In a study by Vassilopoulou-Sellin et al. the incidence of GHD was investigated in 19 poorly growing children with NF1 and without other identifiable risk factors for shortness [7]. Seventy-nine percent (79%) were diagnosed as having GHD on the basis of a pick GH response <10 mcg/L after clonidine stimulation and 42% had a pick GH level <5 mcg/L, indicating a high frequency of profound GHD in this cohort. The causal mechanism of increased frequency of GHD in patients with NF1 remain to be elucidated. It is still plausible that despite the high-resolution capability of current MRI neuroimaging, cerebral abnormalities responsible for GHD are present, but not readily identifiable. Another possible explanation could be that there are abnormalities occurring at the cellular level, implicating the known molecular function of neurofibromin in signal transaction.
In this case report, because we suspected NF, first of all we programmed a study of the genetic framework. At the same time, we detected the deficit of GH and we required a MRI brain scan to conduct a pituitary study, as per protocol.
Finally, we would like to point out that the recombinanthuman- GH (rhGH) therapy, at the usual doses is well tolerated and that current knowledge suggest that such treatment does not influence the progression of any of the features of NF1, including the incidence, of recurrence of primary or the development of secondary intracranial tumors. Although these observations on a substantial number of patients are reassuring, there remains the problem of GH treatment in subjects with inherent risk of malignancy [8].
Conclusion
Our case was unusual in presenting with an unusually aggressive course at younger age with involvement of the optic radiations requiring radiotherapy for the same. We would like to reiterate the importance of a thorough systemic evaluation of any child presenting with short stature and other symptoms suggesting a systemic disease.
18731
References
- Vassilopoulou-Sellin R (1996) Precocious puberty, growth hormone deficiency, and neurofibromatosis. J Pediatr. 128: 166.
- Créange A, Zeller J, Rostaing-Rigattieri S (1999) Neurological complications of neurofibromatosis type 1 in adulthood. Brain 122: 473-481.
- de Winter AE, Moore BD, Slopis JM, Ater JL, Copeland DR (1999) Brain tumors in children with neurofibromatosis: Additional neuropsychological morbidity? Neuro Oncol 1: 275-281.
- Frindik JP (2001) Pituitary morphologic anomalies and MRI in pediatric growth hormone deficiency. The Endocrinologist 11: 4.
- Theos A, Korf BR (2006) Pathophysiology of neurofibromatosis type 1. Ann Intern Med 144: 842-849.
- Cnossen MH, Stam EN, Cooiman LC, Simonsz HJ, Stroink H, et al. (1997) Endocrinologic disorders and optic pathway gliomas in children with neurofibromatosis 1. Pediatrics 100: 667-670.
- Vassilopoulou-Sellin R, Klein MJ, Slopis JK (2007) Growth Hormone deficiency in children with neurofibromatosis 1 without suprasellar lesions. Pediatric Neurology; ISSN 0887-8994.
- Howell SJ, Wilton P, Shalet SM (1998) Growth hormone replacement and the risk of malignancy in children with neurofibromatosis. J Pediatrics 133: 201-205.