Keywords
|
| mTOR; Epileptogenesis; Rapamycin; Gliosis; Inflammation factors |
Introduction
|
| Epilepsy affects about 1% of people and is associated with significant morbidity and mortality. Approximately one-third of epileptic patients are intractable to currently available treatments [1]. In responsive cases, medications can suppress seizures symptomatically, but there is minimal evidence that existing “antiepileptic” drugs correct the underlying brain abnormalities causing epilepsy (epileptogenesis) or alter the natural history of epilepsy. Therefore, a major objective is to develop a treatment that could prevent epilepsy in patients with high risk of developing epilepsy [2,3]. To develop such treatments, a better understanding of the cellular and molecular mechanisms of epileptogenesis is required. |
| Brain inflammation has been strongly implicated in the pathophysiology of epilepsy [4,5]. While activation of inflammatory mechanisms in response to acquired brain injury is perhaps not surprising, accumulating evidence suggests that brain inflammation is an important mechanism that promotes epileptogenesis and seizure development [6]. In fact, inflammatory markers, such as cytokines and chemokines, have been found in brain specimens from patients with cortical malformations, including TSC [7,8]. Altered mTOR signaling is observed in a variety of cortical malformations and neurodegenerative diseases [9,10]. Several of these disorders, including Tuberous Sclerosis Complex (TSC), focal cortical dysplasia, hemimegalencephaly, and ganglioglioma are highly associated with epilepsy [11], suggesting that mTOR signaling is involved in epileptogenesis. |
| Recently, rapamycin (RAP), the inhibitor of mammalian target of rapamycin (mTOR) pathway, appears to be a drug that can significantly reduce, and in some instances even prevent the development of epilepsy in animal models [12-14]. Increasing evidence also implicates mTOR dysregulation in the pathogenesis of acquired forms of epilepsy, such as infantile spasms (IS) and temporal lobe epilepsy (TLE). Althoughthe obtained neuroprotective effect suggests that brain inflammation might also be reduced by RAP, this has not been investigated until now. It has been reported that RAP reduces brain inflammation after brain injury such as traumatic brain injury and focal ischemia in rat [15,16]. |
| While numerous downstream mechanisms may mediate epileptogenesis, less is known about initial signaling pathways that trigger the subsequent changes in the brain causing epilepsy. We hypothesized that the effect of RAP on seizure development might be achieved by an antiinflammatory action. To test this we determined the extent of status epilepticus (SE)–induced brain inflammation after chronic RAP treatment in kainic acid induced SE rat. Activation of astroglia and microglial, and high expresson of inflammation factors (such as inter leukin-1β (IL-1β), cyclooxygenase-2(COX-2) and transforming growth factor-1(TGF-1)) were considered as markers of inflammation. First we studied the alteration of mTOR signal pathway after Kainic acid induced SE, and next investigated the effects of RAP on seizure development. Also we studied the effects of RAP on several early inflammatory responses after KA induced-SE such as the activation of astrocytes and microglias, and activation of inflammation factors. Finally, we correlated these potential proepileptogenic mechanisms with the development of post-SE epilepsy and seizures after RAP treatment. |
Materials and Methods
|
|
Experimental animals
|
| 10 days post-birth immature male Sprague-Dawley rats (Experimental Animal Center of Wuhan University, Wuhan, China) weighing 30–50 g were used in this study, which was approved by the university animal welfare committee. All efforts were made to minimize animal suffering and reduce the number of animals used in each series of experiments. The rats were housed individually in a controlled environment (21 ± 1°C; humidity 60%; lights on 08:00 AM to 8:00 PM; food and water available ad libitum). |
| Animals were randomized to treatment groups, drugs were delivered via an intraperitoneal injection in 0.3 mL volume, 1 h after KA injection. Phenobarbital was used at 30 mg/kg to terminate the seizures. |
|
Status epilepticus induction
|
| Status epilepticus (SE) was induced by intraperitoneal injection of kainic acid (10 mg/kg, Abcam). The behavior of the animals was observed, and seizures were scored according to the Racine scale. One hour after the onset of SE (Class V on the Racine scale indicated by rearing followed by falling down as seen for a full motor seizure), seizures were terminated by subcutaneous injection of sodium pentobarbital (PB; 30 mg/kg). Control rats received the appropriate volume of saline vehicle followed by PB 1 h later. |
|
Continuous video monitoring
|
| Kainic acid-injected rats (KA) were video monitored from day 2 to day 21 post-SE and were scored from 7 am to 3 pm or from 4:30–7 pm by an individual blind to the condition. Behavioral seizures ranging from Class III (dog shake) to Class V (full rearing followed by falling down) on the Racine scale were scored. |
| Following a midline scalp incision, three partial cranial holes were drilled on the skull surface for placement of Electroencephalogram (EEG) screw electrodes (Bilaney Consultants Ltd, Sevenoaks, UK). A complete craniectomy was drilled for the placement of a guide cannula for the intra-amygdala injection. Coordinates from bregma were AP: −1.4 mm and L: +2.9 mm. The guide cannula was lowered to rest on the brain surface and the entire assembly fixed with dental cement. Pups were then placed in an incubator at nest temperature (35 ± 1°C) to recover from surgery for 30 min prior to further experimentation. |
|
Rapamycin treatment
|
| For rapamycin (LC Laboratories) injection, we followed a modified protocol reported by Meikle et al. [17]. Rapamycin was dissolved at 50 mg/ml in ethanol and stored at − 20°C. Before each administration, rapamycin was diluted in 7% Tween 80, 5% polyethylene glycol 400 (PEG-400) and water to final 6% ethanol. Rapamycin or vehicle (7% Tween 80, 7% PEG-400, 6% ethanol) was given at 6 mg/kg intraperitoneally [18]. Pretreatment of rapamycin was given intraperitoneally (6 mg/kg/day under isoflurane anesthesia) for 3 consecutive days prior to kainate injection, once daily for 7 days (n=20). This specific time point was chosen, rather than shortly after SE, since we wanted to investigate the effects on spontaneous seizures, which require at least 4 h of SE in this animal model [19]. Hereafter RAP was given every other day until rats were sacrificed. In addition, VEH SE rats and RAP-treated controls were included. The dose of RAP was based on previous experiments in epileptic rats in which it has been shown to reduce seizure activity [20]. |
| To examine the expressions of mTOR and inflammatory molecules, rats were divided into three groups: control group, KA group and KA+RAP group. The rats in control group were injected with vehicle solution that contained 5% Tween 80, 5% PEG 400, and 4% ethanol. In KA group, the rats were injected with kainic acid (10 mg/kg). In the KA+RAP group, pretreatment of rapamycin was given intraperitoneally for 3 consecutive days prior to kainate injection, once daily for 7 days. Each group consisted of 20 rats. To investigate the molecular mechanism of mTOR regulation, rats were sacrificed at 1 h, 3 h, 8 h, 24 h, 3 d, 1 w, 2 w, 6 w after SE. In addition, to examine the mechanism of inflammations, rats were sacrificed at 1 w and 2 w after SE. Rats in the control group were only injected with an equal volume of dissolvent. Rats were sacrificed by decapitation and hippocampi were removed and stored at −80°C. |
|
Immunocytochemistry
|
| Rats were deeply anesthetized and perfused transcardially first with 100 ml 10 mmol/L PBS (pH 7.4) and then with 100 ml fixative (4% (w/v) paraformaldehyde, 15% (w/v) saturated picric acid, and 10 mmol/L PBS). The brains were removed and post fixed in fixative for 1 d. Coronal blocks (3 mm) were cryoprotected overnight in 30% (w/v) sucrose. We removed the brains and either cryopreserved tissue blocks for cryostat sectioning (30 μm) or embedded them in paraffin for sections (7 μm). The coronal sections through the dorsal hippocampus (at the level corresponding to 2.8~4.5 mm posterior to bregma) were selected, and each one-seventh section of the hippocampus (6 sections per animal) was examined for quantitative immunohistochemical analysis. Immunohistochemistry was carried out as described previously. The specimens were fixed in 4% (w/v) paraformaldehyde in PBS (pH 7.5) for 40 min, washed with PBS, incubated for 2 h with blocking buffer (2% (w/v) horse serum/1% (w/v) bovine serum albumin (BSA)/0.1% (w/v) Triton X-100 in PBS, pH 7.5), then incubated with primary antibodies overnight at 4°C and with secondary antibodies for 1 h at room temperature. Primary antibodies used as markers for the activation of the mTOR pathway were rabbit anti-murine polyclonal antibody to p-S6 (1:300, Santa Cruz, Inc., USA); glial fibrillary acidic protein (GFAP, 1:200, Santa Cruz, Inc. USA) for the markers of the activation of astrocyte; for the mouse anti-rat OX-42 antibodies (1:500, Chemicon, USA) as a marker of microglia. Biotinylated goat antirabbit IgG (ABC, Sigma, USA) and anti-mouse IgG (1:100, Biodesign, USA) or tetramethylrhodamine isothiocyanate (TRICT)- conjugated anti-mouse antibody were used as the secondary antibodies. Double immunostaining used primary antibodies directed to monoclonal anti-BrdU (1:300, Pharmingen, USA) and TRICT-conjugated secondary anti-mouse antibody to label newly generated cells. GFAP and an anti-mouse fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:100, Vector Laboratories, USA) were used to label astroglial cells. The colocalization was analyzed using a laser scanning confocal microscope with a Bio-Rad MRC 1024 (argon and krypton). The fluorescence signals were detected at excitation/emission wavelengths of 550/620 nm (TRICT, red) and 488/522 (FITC, green), respectively. Biotin signals were detected with 3,3’-Vdiaminobenzidine (DAB, brown) |
|
RNA isolation and real-time PCR
|
|
RNA Extraction:
|
| For isolating the hippocampal RNA, the brains of the animals were removed, the hippocampus was dissected, placed in sterile tubes containing 1 ml of the Trizol Reagent (Invitrogen Life Technologies, USA), and frozen at −80°C until used. Total RNA extraction was performed after hippocampus homogenization (15 strokes with a Thomas Scientific AA Teflon homogenizer) according to the manufacturer’s instructions. The RNA samples were suspended in 50 μl of nuclease-free water. The concentration and purity of RNA were determined at 260/280 nm using a nanodrop spectrophotometer (Ocean Optics, Dunedin, FL, USA). The integrity of the total RNA was assessed by agarose gel electrophoresis, using ethidium bromide staining. |
|
cDNA Synthesis:
|
| The cDNA was obtained by reverse transcription of total RNA, using the kit Super Script III First-Strand Synthesis SuperMix (Invitrogen Life Technologies, USA). For that purpose a small aliquot containing 2 μg of total RNA, suspended in nucleasefree water was mixed with 0.5 μl oligo (dT)20 (50 μM), 0.5 μl annealing RAPbuffer, and brought up to a final volume of 4 μl with nuclease-free water. This mixture was incubated at 65°C for 5 min and then chilled with ice. For reverse transcription, 1 μl of III/RNaseOUT™ enzyme mix and 5 μl of the 2X first-strand reaction mix (10 mM MgCl2, and 1 mM of each dNTP) were added before incubation at 50°C for 50 min. The reaction was stopped by heating the mixture at 85°C for 5 min. The cDNA resulting from this procedure was stored at −20°C until use. |
|
Polymerase Chain Reaction (PCR):
|
| The effect of Rapamycin on the IL-1β, COX-2 and TGF-1 mRNA expression induced by KA was evaluated by reverse transcription-polymerase chain reaction (RTPCR) using the kit GoTaq DNA Polymerase. Briefly, 1.5 μl of cDNA (250 ng/μl) were amplified in a mixture containing 2 μl of 5X green buffer, 0.8 μl of MgCl2 (25 mM), 0.25 μl of PCR nucleotide mix (10 mM), 0.5 μl of the sense primer (10 pM), 0.5 μl of the antisense primer (10 pM), 0.05 μl of DNA Polymerase (5 u/μl) and 4.4 μl of sterile Milli-Q water. |
| PCR reactions were done in an Eppendorf Mastercycler gradient (USA). The temperature cycling conditions were: initial denaturation at 95°C for 5 min, followed by 34 cycles, including denaturation at 94°C for 30 s, primer annealing for 45 s at 58°C for IL-1β and β-actin or at 66°C for TNF-α, and primer extension at 72°C for 1 min. A final primer extension was performed at 72°C for 10 min after which the samples were immediately cooled at 4°C. The nucleotide sequences for the IL-1β primers were: AATGACCTGTTCTTTGAGGCTGAC (sense) and CGAGATGCTGCTGTGAGATTTGAAG (antisense); and the expected product size were 519 bp. The nucleotide sequences for the COX-2 primers were: CCATCCTGGAAAAGTCGAAGTTTAT (sense) and TTTGCCCAGCACTTCACTCATC (antisense); and the expected product size were 663 bp. The nucleotide sequences for TGF-1 primers were: CGTTCGATGTTCGAAGC (sense) and GCCCTCGAAGGTGTTG (antisense); and the expected product size were 536 bp. The nucleotide sequences for the β-actin primers were: TGCTGGTGCTGAGTATGTCGTG (sense) and CGGAGATGATGACCCTTTTGG (antisense); with the expected product size of 302 bp. |
| β-Actin was used as a control to normalize the relative mRNA amount of the amplified cytokines. A negative control in the absence of sample was run together with each transcript. |
| PCR products were separated by 1.5% agarose gel electrophoresis at 90 volts, stained with ethidium bromide and the resulting bands were quantified by densitometry using a MiniBIS Pro Gel Documentation System (Bio-America, Miami FL, USA) and ImageJ version 1.42 software (National Institute of Health, USA). Results are expressed as relative mRNA level expression (IL-1β/β-actin, COX-2/β-actin or TGF-1/β-actin ratio). |
|
Western blot analysis
|
| Hippocampi tissues were homogenized in buffer (25 mM Tris–HCl pH 7.5, 1 mM EGTA, 2 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 10 mM sodium pyrophosphate, 0.2% NP-40, 0.1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol and centrifuged at 12,000×g at 4 °C for 10 min. The supernatants were collected as cell proteins. The concentration of protein was determined by using the BCA protein assay (Pierce, USA). |
| 40 μg of protein were separated by sodium dodecyl sulfatepolyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. Later it was blocked in 5% fatfree milk in TBST buffer for 1 h, the membranes were incubated with primary antibody against phospho-S6 (1:1000 dilution; Cell Signaling Technology, USA), IL-1β, TGF-1 and COX-2 (1:500; Santa Cruz Biotechnology, CA, USA) and β-actin (1:4000; Santa Cruz Biotechnology, CA, USA) at 4 °C overnight. Then the membranes were washed and incubated with horseradish peroxidase conjugated secondary antibody (1:3000; Santa Cruz Biotechnology) for 1 h. Immunoreactivity was enhanced by chemiluminescence kit (Pierce, Rockford, IL, USA) and exposed to film. The band intensity was analyzed with an image analyzer (Alpha Innotech 2200, San Leandro, CA, USA). |
|
Statistical comparisons
|
| All data are expressed as mean ± S.E.M. Statistical comparisons were made using one-way or two-way ANOVA followed by Tukey– Kramer or Dunnett’s multiple comparisons post-hoc test or Student’s t-test with Origin 10.0 software (GraphPad). Data were considered statistically significant if p<0.05. |
Results
|
|
The incidence of spontaneous recurrent seizures (SRS) in chronic periods
|
| Ninety rats injected with kainic acid developed SE which was characterized by continuous motor limbic seizures accompanied by intermittent rearing and falling with a mean latency of (10 ± 2) min. The duration of SE was controlled at 60 min. After the rats were sacrificed at each time point, 20 rats in KA group were monitored in the chronic periods (28 days to 42 days after SE), and 14 mice developed SRS. SRS occurrence rate was 70%. 8 mice of 21 pups in KA+RAP group have observed to SRS, the occurred rate was 38.1%. In Control group, none of SRS was observed. Rapamycin intervention significantly reduced incidence of SRS in chronic phase (P=0.04; chi square test) |
|
The activation of the mTOR pathway after SE and Rapamycin pretreatment blocks kainate seizure-induced mTOR activation
|
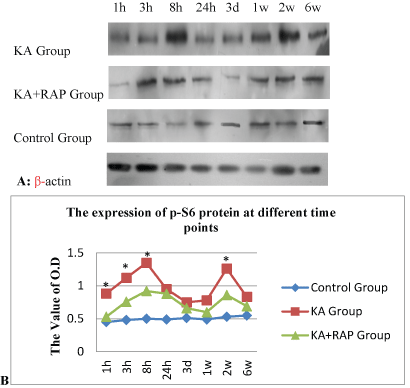
| We determined whether the mTOR pathway was activated after KA induced SE. The expression of p-S6 was the marker of the activation of mTOR signal, and was assayed by Western blotting at different time intervals after seizure onset. Kainate induced-SE resulted in a biphasic activation of the mTOR pathway, including both acute and chronic periods of increased P-S6 expression correlating with acute seizure activity and chronic epileptogenesis, respectively (Figure 1). In the acute phase, a significant increase of P-S6 was initiated within 1 hour of the onset of kainate-induced seizures. The increase in mTOR activity peaked around 8 hours and then decreased to the baseline after 24 hours. Following kainate status epilepticus, mTOR activity remained at pre-seizure baseline for at least 3 days, but then subsequent assays of P-S6 expression revealed a second period of mTOR activation, starting approximately 1 week after kainate injection (Figure 1A). This increase in mTOR activity reached a maximum by about 2 weeks and gradually decreased over the following few weeks, returning to baseline by 6 weeks after kainate status epilepticus. |
| We attempted to block the kainate-seizure induced mTOR activation with the mTOR inhibitor, rapamycin. We found that pretreatment of rapamycin at a dose of 6 mg/kg/day i.p. for 3 consecutive days prior to kainate injection was effective in preventing the initial increase in P-S6 activated over several hours by KA-induced seizures. Similarly, rapamycin pretreatment almost completely blocked the delayed increase in P-S6 over several weeks in hippocampus. RAPbuffer treatment can significantly reduce the expression of P-S6 and reverse the activation of mTOR (Figure 1B). |
|
The expression of p-S6 positive cells in hippocampus and cortex at 2 week after SE
|
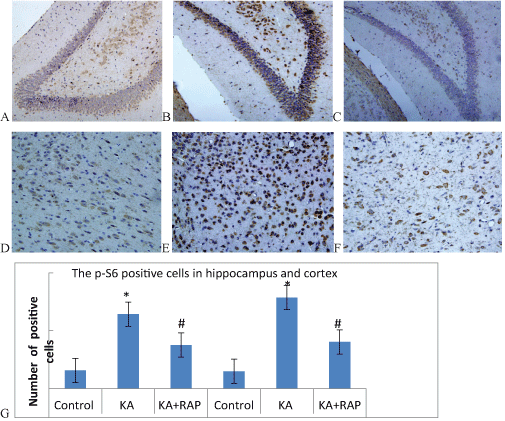
| To assess the extent of mTOR activation in the chronic phase, we also performed immunostaining of p-S6 in sections of 2-week post-SE rats. The p-S6 was expressed in both cytoplasm and nucleus, and it can be seen that the brown particles precipitate in the nucleus periphery of the neurons in the hippocampus and cortex both in the Control Group and KA Group (Figure 2A-2F). At 2 Weeks after KA-induced SE, the expression of positive cells in hippocampus was (15.7 ± 1.6) in Control Group; (64.3 ± 6.9) in KA Group and (37.6 ± 3.7) in KA+RAP group. The difference was statistically significant. The expression of positive cells in cortex was (14.9+2.0) in Control Group; (78.7+5.5) in KA Group and (40.3+4.7) in KA+RAP Group. The difference was also statistically significant (Figure 2G). RAP-treatment in post-SE rats the p-S6 immune positive cells was decreased comparing with KA group, and similar to control values, indicating that RAP treatment could attenuate seizure induced overexpression of p-S6 and that RAP treatment was effective in inhibiting the mTOR pathway in our animal model. In KA-treated epileptic rats, a significant increase of p-S6 IR was observed in neurons at hippocampal and cortex compared to control Group (Figure 2G). RAP-treatment effectively inhibited neuronal expression of p-S6 as shown by the strongly decreased staining comparing with KA Group, although it was still somewhat higher than in controls. |
|
Rapamycin reduces the activation and proliferation of astrocyte at 1 week after SE
|
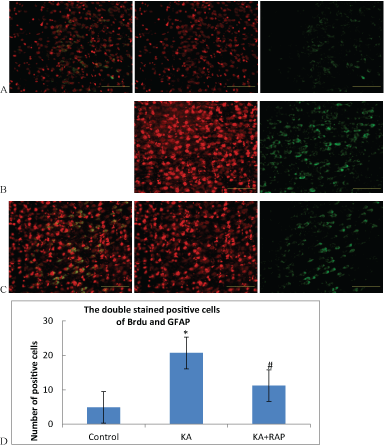
| BrdU labeled as the marker of new cells, GFAP labeled as the marker of astrocytes. Double stain of BrdU and GFAP labeled as the marker of the proliferation and activation of astrocytes. The number of positive cells in the Control Group was (5.7+1.25). The number was (30.4+2.3) in the KA Group and (18.4+3.4) in the KA+RAP Group. The expression differences in each group were statistically significant (Figure 3). After KA-induced SE, the activation of astrocytes were increased significantly, and rapamycin intervention inhibited the activation of mTOR and decreased the proliferation and activation of astrocytes. |
|
The activation of microglias at 1 day after KA-induced SE
|
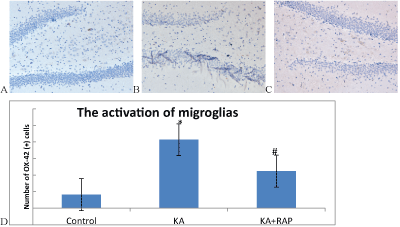
| Microglias(MG) are normally resting state in normal rat brain, anti- OX-42 immunohistochemical staining is negative in the slice and is not easy to find. MG cell morphology is not clear (Figure 4A). The first days after SE, microglios were activation state, OX-42 stained-positive cells were clear in cell morphology, cell body becomes large, and the projections can be seen on the small spines (Figure 4B). In Rapamycin treatment group, the number of immunoreactive cells reduced (Figure 4C). The number of OX-42 positive cells in Control Group was (4.1 ± 1.29) and (20.7 ± 1.77) in KA group and (11.2 ± 1.23) in KA+RAP group. The differences among the groups were significantly (p<0.001) and RAP treatment inhibited the activation of microglias (Figure 4D). |
|
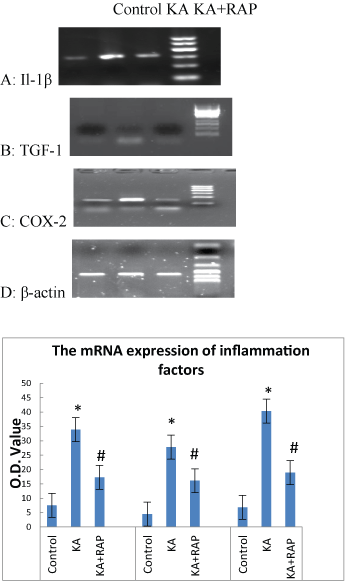
The alteration of early inflammatory molecules (such as L-1β, TGF-1 and COX-2) mRNA expression in each group
|
| We further explored the effects of mTOR signal inhibition on the expression of these three inflammatory molecules. We quantified the mRNA levels of IL-1β, TGF-1, COX-2 in rat hippocampus at 1 h after SE. As shown in Figure 5A, the value of mRNA level of IL-1β in Control Group was (7.5 ± 1.08), the mRNA level rised (33.9 ± 1.19) at 1 h after SE in KA group and was higher than in the KA+RAP group (17.3 ± 2.05) (P<0.05) (Figure 5D). The expression of TGF-1 mRNA in Control Group was (4.5 ± 1.08) and increased to (27.8 ± 1.31) at 1 h after SE and was higher than in the KA+RAP group (16.1 ± 1.19) (P<0.05) (Figure 5B and 5D). In addition, the mRNA level of COX-2 increased at 1 h after seizures (40.7 ± 2.27), and was higher than in the KA+RAP group (19.1 ± 2) (P<0.05) (Figure 5C and 5D). We found that mRNA expressions of IL-1β, TGF-1, COX-2 in rat hippocampus at 1 h after SE increased significantly and rapamycin suppressed the expressions of these early inflammatory molecules (Figure 5D). |
|
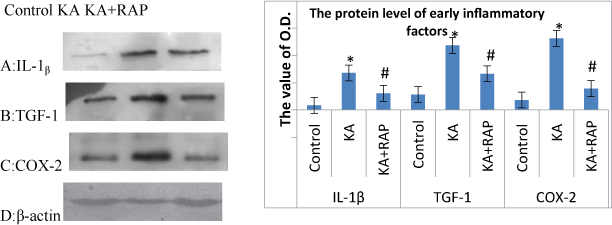
The alteration of inflammatory molecules (such as IL-1β, TGF-1 and COX-2) protein level at 24 h after SE in each group
|
| We also investigated the expression of protein levels of IL-1β, TGF1, COX-2 in rat hippocampus at 24 h after SE as shown in Figure 6. The levels of IL-1β, TGF-1, COX-2 all obviously increased at 24 h after KA treatment. The Rapamycin and KA co-administration significantly decreased the expressions of IL-1β, TGF-1, COX-2 (P<0.05) (Figure 6A-6D). |
Discussion
|
| The main findings of this study can be summarized as follows: mTOR pathway was activated after KA-induced SE, chronic RAP treatment starting 3 days before KA- induced SE, strongly reduced the development of epilepsy. RAP treatment also reduced SE-induced activation of astrocyte and microglios in hippocampal, and reduced the expressions of early inflammatory factors such as IL-1β, TGF-1, COX-2. |
| TSC is caused by mutations in the TSC1 or TSC2 genes, which leads to hyperactivation of the mammalian target of rapamycin complex 1 (mTORC1) pathway. The mTOR, a 290 kD serine–threonine protein kinase, activation of mTOR results in phosphorylation of ribosomal protein S6 kinase and eukaryotic initiation factor 4 binding proteins, which increases a subset of mRNA translation and protein synthesis [21]. Recent studies have showed that mTOR signaling is abnormally activated by seizures and the mTOR is associated with epileptogenesis [20,22-24]. In kainate induced SE model, there is an acute transient mTOR activation phase lasting for several hours and a late longer phase lasting for several weeks in rat hippocampus [24]. Rapamycin pretreatment prior to SE is able to block both the phases of mTOR activation and correspondingly decrease the frequency of spontaneous seizures [24]. Our study also showed that the mTOR pathway was upregulated after acute convulsions induced by KA administration. Meanwhile, Rapamycin pretreatment could inhibit mTOR activation and modulate the following inflammations. mTOR inhibitors are being tested in clinical trials as potential treatments for epilepsy [25]. But the critical mechanisms of mTORC1 inhibitors anti-epilepsy are poorly understood. Identification of these downstream mechanisms may lead to more targeted therapies, with more specific efficacy. |
| The molecular mechanisms that lead to the development of epilepsy following an initial precipitating injury of the brain are incompletely understood. In the present study, we implicate involvement of the mTOR signaling pathway in mediating epileptogenesis in a rat model of temporal lobe epilepsy following an episode of status epilepticus. We also demonstrated that the mTOR pathway is strongly activated with a biphasic time course after kainate-induced status epilepticus, and this mTOR activation could be blocked by rapamycin treatment. Rapamycin pretreatment counteracted known effects of kainate status epilepticus on the activation of gliosis, neurogenesis, and inflammation, which may represent cellular mechanisms causing epileptogenesis in this model. Pre-treatment with rapamycin significantly decreased the development of chronic epilepsy, indicating that mTOR signaling is involved in epileptogenesis and that mTOR inhibitors could be useful as “anti-epileptogenic” therapy. |
| In the present study, the initial injurious stimulus of status epilepticus leads to a biphasic activation of mTOR, with the first peak of activation occurring in hippocampus acutely within a few hours during the seizure activity and delayed peak occurring several days later within hippocampus. Since mTOR can be triggered by glutamate receptor stimulation [26,27], the initial widespread mTOR activation occurs with status epilepticus, which causes massive glutamate release. Both the temporal and spatial distribution of mTOR activation suggests that mTOR could be central to epileptogenesis in this model. Furthermore, as mTOR inhibition by rapamycin attenuated the development of epilepsy and reduced the inflammation responses after SE implicated in epileptogenesis in the kainate model, these data support that mTOR plays a primary causal role in epileptogenesis and may serve as an initial central signaling trigger that activates multiple downstream epileptogenic pathways and processes. |
| Proinflammatory mechanisms have been implicated in contributing to the pathophysiology of epilepsy. The role of brain inflammation in the pathophysiology of various types of epilepsy has received increasing attention, especially in epileptogenic brain injuries [4,5,28]. Different early inflammatory factors, such as cytokines and chemokines, are activated in a variety of animal models of epilepsy, including chemoconvulsant and electrical kindling models of epilepsy, as well as in human tissue obtained from epilepsy patients with mesial temporal sclerosis [29-32]. Furthermore, anti-inflammatory treatments targeting these pathways have begun to be explored. For example, selective pharmacological inhibition of the cytokine IL-1β production in astrocytes inhibits seizures in rats [33,34]. Although similar inflammatory markers have also been found in tubers from TSC patients [8,35,36], the pathophysiological significance of inflammation in TSC, such as for epilepsy is unknown. Whether the mTOR inhibitors could prevent the activation of the inflammation after seizures and anti-epileptogenesis in chronic phase is poor understood. |
| Based on recent literature and the immunosuppressant properties of RAP, we expected to find a reduction of astrocyte and microglia activation and inflammation after the treatment. This would have helped to explain RAP’s suppressing effect on epileptogenesis after SE, which, according to the hypothesis, can be modulated by antiinflammatory treatment [6]. Recently, it has been reported that RAP produces a neuroprotective and antiinflammatory effect in the brain in a focal cerebral ischemia rat model [16]; moreover, microglia activation is reduced by RAP in a closed head injury mouse model together with an increased neuroprotection [15]. We detected the inhibitor of microglia activation by RAP in the brain. Similarly, the suppression of the activation and proliferation of astrogliosis was still evident after SE in RAP-treated rats. RAP treatment in mouse models for TSC reverses the histologic abnormalities, including astrocytosis [12]. We also find the evidence that RAP decreased the number of activation and proliferation of astrocytes. Despite the suppressing effect of RAP on parenchymal inflammation and gliosis, seizure development was strongly reduced. These data support the involvement of brain inflammation or gliosis as epileptogenic mechanism, and we can presume that the mTOR inhibitors specific inflammatory pathways. |
| Studies have revealed that seizures may activate inflammatory cytokines and proinflammatory molecules [29]. The enhanced production of inflammatory cytokines such as COX-2, TGF-1 and IL-1β may directly regulate neuronal excitability and other processes involved in epileptogenesis [29,33,37]. Inflammatory cytokines may contribute to altered neuronal network excitability during seizures and consequently affect the downstream events involved in epileptogenesis. Recently, IL-1β is proved to inhibit epileptogenesis by regulating the synaptic transmission and neuronal plasticity via neuronal sphingomyelinase [38]. In this study, we observed that SE induced by KA increased the expressions of COX-2, IL-1β, TGF-1 in rat hippocampus in a time dependent fashion. The findings from our study support a potential anti-inflammation role of mTOR inhibitors after KA induced SE. We also demonstrated that rapamycin was able to reverse the inflammatory molecule expressions. |
| Studies have indicated that rapamycin administration can reduce the incidence of convulsions [39-41]. Our results showed that rapamycin treatment prior to kainic acid could reduce the development of SRS. Our present study indicated that the regulation of inflammations induced by seizures may also be associated with the neuroprotection of rapamycin. |
Conclusion
|
| RAP treatment leads to a suppression of seizure development in the rat TLE model. This suppression is linked to a containment of astrocytes and Rapamycin inhibits overexpression of inflammatory cytokines including IL-1β, TGF-1 and COX-2 activation via mTOR signaling after seizures. Rapamycin is a promising therapeutic agent against seizures. |
Acknowledgments
|
| This work was supported by grants from the Renmin Hospital of University (RMFH 0001) . |
Figures at a glance
|
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
 |
 |
 |
| Figure 4 |
Figure 5 |
Figure 6 |
|


