Keywords
novel Corona Virus (2019nCOV); Spike glycoprotein; Fusion peptide; Lead Molecules; Fusion inhibitor
Introduction
Outburst of the novel corona virus that appear within the central Chinese city of Wuhan in late December are being sign on daily round the world. Quite 3,23,256 people have died from (COVID-19), the disease originated by the novel corona virus, while some 4.9 million infections are reported in additional than 185 countries and territories [1]. Approximately quite 1.3 million people have recovered so for. Severe acute respiratory syndrome corona virus (SARS-CoV) rose in humans from an animal reservoir in 2002 and swiftly spread globally causing 8,096 cases and 774 associated mortality in 26 countries through July 2003 [2]. SARS CoV again in a very small epidemic in 2004, but has since vanish from human circulation. Like severe acute respiratory syndrome (SARS)-CoV, COVID-19 similar to lineage B β-corona virus, and it’s the aptitude to acknowledge human cell surface protein angiotensin-converting enzyme-2 (ACE2) and as a receptor to infect host cells [3,4]. Corona viruses are round shaped or idiomatic, with a diameter of 80-120 nm. While examining under the electron microscope, the virion surface is trim with clublike ledge assembled by the trimeric (mushroom) like spike (S) glycoprotein. The viral capsid is surrounded by the membrane (M) glycoprotein, the foremost abundant structural protein that embeds within the envelope via three transmembrane domains. Supplementary, a small trans-membrane protein called the envelope (E) protein is also present in a very few amounts within the envelope [5-8]. The nucleocapsid (N) protein binds to the RNA genetic material in a very beads-on-a-string fashion, forming the helical symmetrical nucleocapsid. The corona virus genome could be a positive-sense, non-segmented, single-stranded RNA, with startle large size scale from 27 to 32 kb. Corona virus central dogma is initiated by the binding of Spike glycoprotein protein to the cell surface receptor (s). The Spike protein is distinguished of two functional subunits, S1 (bulb) for receptor binding and S2 (stalk) for membrane fusion. Determined interaction between S1 and therefore the cognate receptor depart an extreme conformational change within the S2 subunit, prime to the fusion between the virus envelope and therefore cellular membrane and release of the nucleocapsid into the host cytoplasm. SARS-CoV spike (S) protein S2 subunit plays an essential role in conciliating virus fusion with and entry into the host cell, during which the heptad repeat-1 (HR1) and heptad repeat 2 (HR2) can merged to make six-helical bundle (6-HB), thereby cross-over viral and cellular membranes in close proximity for fusion, whether a COVID-19 Spike-HR1 can also function a vital target for the event of COVID-19 fusion/entry inhibitors [9-11]. The aim of the present study was to estimate the inhibitory and interaction analysis of virus spike glycoprotein (S) with Alangium salvifolium phytoderivatives (Figure 1) [12] by usage of various bioinformatics tools.
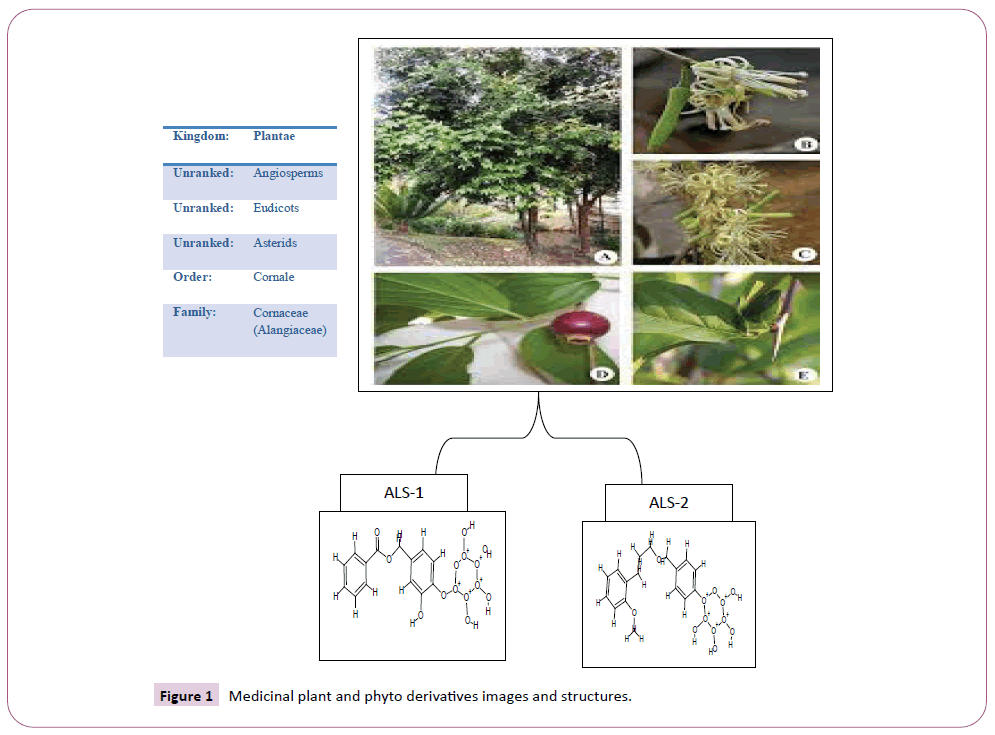
Figure 1: Medicinal plant and phyto derivatives images and structures.
Materials and Methods
Ligand preparation
The lead compounds were collected from an editorial published by Suresh Shravya and his coworkers [13]. The compounds were selected from Salviifoside derivatives from the genus Alangium, especially from the A. salvifolium plant. Chemical structure of Alangium salvifolium plant derivatives ALS-1 and ALS-2 were drawn with the using of CHEM-sketch software with possible structure definition file format for docking (Chem-Sketch-www. acdlabs.com/download/), an absolutely strapping chemical structure drawing program [14]. The chem.-sketch file format.sk1 converted to mol. file for further ligands physical characterization by using Pymol, arguslab, and online pkCSM (http://biosig. unimelb.edu.au/pkcsm/prediction_single) software.
Retrieve receptor protein
Protein Data Bank (PDB) is that the worldwide database of structural and observational data of biological macromolecules, established in Brookhaven National Laboratories. It collected structural and observational data of the macromolecules specially protein data which retrieved by the X-ray crystallographic and NMR methods. 3D structure of trimeric mushroom like virus Spike glycoprotein from PDB, whose PDB ids is 5wrg respectively [15].
Molecular docking and simulation
The protein atoms were typed using the CHARMm field of force. The site of the protein was first identified and defined using an eraser size 10.0 Aº. Molecular docking studies on the above mentioned selected phytoderivatives against virus spike (S) protein (5wrg) was drained in Autodock (vina) in PyRx-phython prescription 0.8, which is freely accessible and designed for molecular docking studies. Autodock-PyRx includes docking sorcerer with an easy-to-use programme which makes it a potential tool for computer-based drug design. Autodock (Vina) PyRx even have facility to chemical spreadsheet-like functionality and powerful visualization engine that are must for cogent drug design [16]. The chosen drug targets were energy minimized, and converted them into pdbqt file format in Autodock-PyRx. Then the ligands were docked into the site of receptor using Pyrx (Autodock) dock procedure. Affinity of binding energy score absolute energy was obtained from the results. Ligand drug potential ability was through with the ARGUSLAB software (ArgusLab-www.arguslab. com/), during which the result is being obtained on the premise of pose energy. Before docking a molecule, first it’s needed to define the atoms that conjure the Ligand like drug, inhibitor, etc., and therefore the Binding Site on the protein where the drug binds. The ultimate results are supported the sort of calculation we run like as Geometry optimization-search for ‘Final Geometry’ and Electronic spectra-search for ‘Excited state properties’
Receptor-ligand binding analysis
Structure of Plant phytoderivatives open with saved pdb file format were upload in Discovery-studio 4.1 version. Use of protein-interaction tool, the binding pattern of receptor and ligands studied and plot the 2-D receptor -ligands graph for amino acid residue and bond formation between receptor pocket molecule and ligand [17]. Auto dock vina generated docking pair of protein and ligands were saved in pdb format, and were visualized in Discovery studio [18] visualization tool i.e. pythonenhanced molecular graphics tool. It excels at three-dimensional visualization of proteins, small molecules, density, surfaces and trajectories. It also includes molecular editing, ray tracing, and movies. The ligand binding sites and surrounding amino acids of ligands were also visualized. Molecular interactions within the sort of hydrogen bonds between proteins and ligands were characterized and therefore the distance of hydrogen bonds was also calculated. Site Prediction proteins have specific sites, the residue side chains that form a full of life cavity or cleft where the ligands or atoms or other proteins are capable to bind and are called active sites.
In-silico pharmacokinetic prediction study
Pharmacokinetic (ADMET) properties that’s Absorption, Distribution, Metabolism, Execration and Toxicity value were examine by using of Admet-SAR and Online swissPort database which provides latest and most inclusive manually created data for various chemicals with known ADMET properties that helpful to pharmacological properties of studied molecule [19]. Predictions of drug properties were calculated in Swissport, an online based pharmacokinetic tool. It determines lipinski rule of 5 (rule of five), an essential to judge drug likeness or determine the chemical compounds pharmacological and biological properties, which concur with the oral prescribed drugs for human [17]. The rule of five values includes cLog P, mass, bond donors and bond acceptor for the drugs. Further, we predicted compounds activity on biological targets (kinase inhibitor, nuclear receptor ligand, protease inhibitor, and enzyme inhibitor)
Results and Discussion
There were various crystal structures cited within the Protein Data Bank (PDB) (www.rcsb.org/pdb) on Spike trimer protein. During this study (PDB ID: 5 wrg) with 2.1 Å resolution Spike glycoprotein trimer (mushroom like) protein structure (Figures 2a, 2b and 2c) molecular docked with Phyto-derivatives of Alangium salvifolium. The reference ligands were docked into the pocket site of the Spike glycoprotein (S) assembly, and also the docking score was found in rage of -11.6 Kcal/mol and -10.8 kcal/mol with % RMSD value (Table 1). The ultimate trajectory files were taken for calculating the receptor-ligands of the complex structures. At the identical time as running receptor-ligand interaction analysis. 2D-Diagram plot shows the steadiness of the complex structures. Interaction analysis clearly marked that phytoderivatives intercept region of for fusion peptide region and HR-1, and interlock between all monomer (Table 2 and Figure 3). previous study the functional domain in (SARS)-CoV, Spike protein, counting N-terminal domain (14-305aa),receptor-binding domain (319-541aa), and receptor-binding motif (437-508aa) in S1 subunit which including 14-685aa and fusion peptide (788-806aa), Heaped region HR-1 (912-984aa), HR2 (aa1163-1213aa), trans-membrane domain (1214-1237aa) and cytoplasm domain (1238-1273aa) in S2 subunit range between 686-1273 residue (Figure 2e). In the post-fusion hairpin conformation of the SARS-CoV or MERS-CoV S protein, the HR2 domain forms both rigid helix and elastic or versatile loop to interact with HR1 region [19]. There are many well built interactions between HR1 and HR2 domains inside the helical region, which is thus designated “fusion core domain” (HR1 and HR2core domain, respectively). Conforming to the sequence alignment, the COVID-19 and SARS-CoV S2 subunits are highly conserved, with 92.6% and 100% overall identity in fusion core domain. However, inside the HR1core region, out of 21 residue only 8 residue show mutation (~38% diversity). This is significantly different from the HR1core region of previously identified SARS-like viruses, like WIV1, Rs3367, and RsSHC014, which are 100% clone of that of SARS-CoV [20]. These novel point mutations in COVID-19, S2 subunit may change the interaction pattern between fusion core domains within the post-fusion core, thus affecting the helical bundle (6-HB) construction supported our past experience. But Alangium salvifolium phyto-molecule bind diversely monomer_A (738-976) monomer_B (951-988) and monomer_c (741-987) that cover fusion peptide to fusion core domain (FP, HR1) derived peptides region, respectively (Table 3 and Figure 3) and explored their bond interaction characteristics. It gives more satisfaction just in case of mutation because studied molecule bind altogether three monomer give more surety to inhibit the targeted region. Since the fusion core domain and S-Hepated region residue 100% inhibit /block, with Alangium salvifolium phytoderivatives may act as a fusion inhibitor in much the identical way as reported SARS-CoV fusion inhibitor [4,20,21]. These result confirm, for the primary time, that ALS- 1 and ALS-2 are ready to interact with fusion peptide region and HR-1 conserved region that affecting the 6-HB formation therefore inhibit COVID-19 fusion with the host cell, as past studies confirmed that in SARS-CoV, MERS-CoV, and other HCoV [4,5,20,21]. When S1 subunit recognizes its receptor on host cell, the fusion core domains are exhibited and merged with one another, forming 6-HB to participate in endocytosis initiation between virus and host cell [22]. Notably, both ALS-1, ALS- 2 fusion core inhibitor, exhibited potent inhibitory activity against Spike protein involved endocytosis. Phytoderivatives which play a pivotal role to inhibit fusion core domain may have a possible to drug against COVID-19. Next step to optimization of drug development step pharmacological evaluation prediction required for qualifying active pharmacological ingredients (API). Therefore, Pharmacological evaluation of phyto-derivatives ALS- 1 and ALS-2 were evaluated for ADMET (Adsorption, Distribution, Metabolism, Excretion and Toxicology) properties and drug likeliness. Both were found to best interaction with virus Spike glycoprotein and violate the Lipinski’s rule. Both phyto-derivatives were screened on the premise of BBB permeability, GI absorption, with optimum solubility, Toxicity and carcinogenic tests screened the compounds and ultimately, both phyto-derivatives, drug-like, having suitable ADMET depicted in Figures 4 and 5.
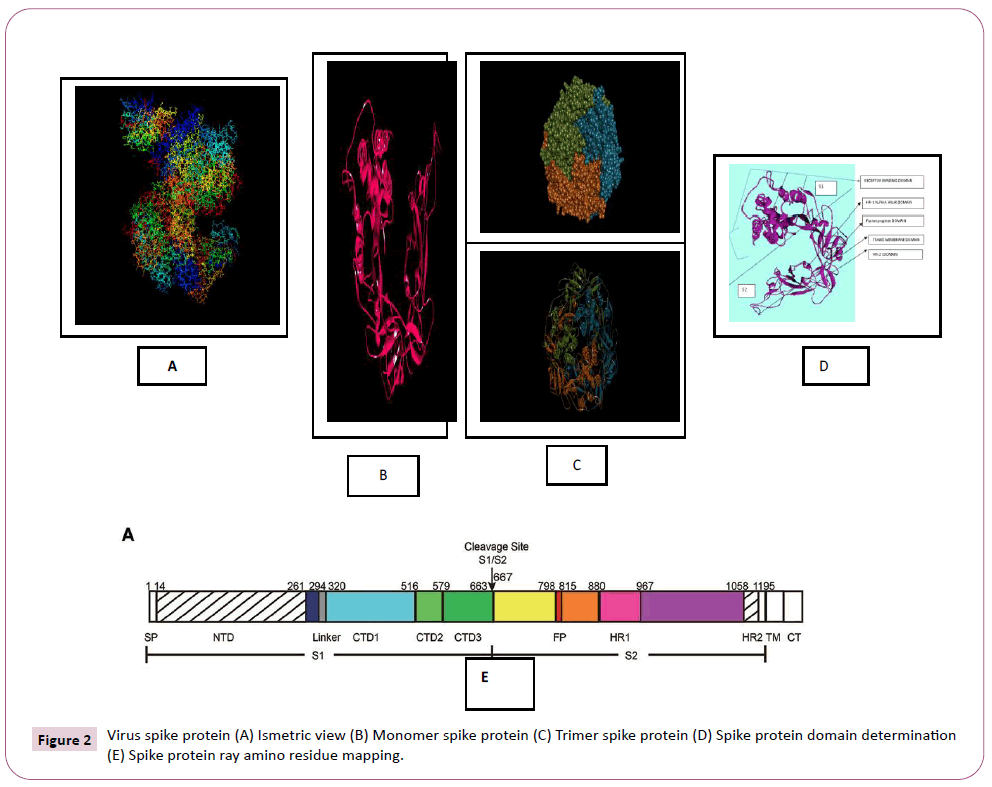
Figure 2: Virus spike protein (A) Ismetric view (B) Monomer spike protein (C) Trimer spike protein (D) Spike protein domain determination (E) Spike protein ray amino residue mapping.
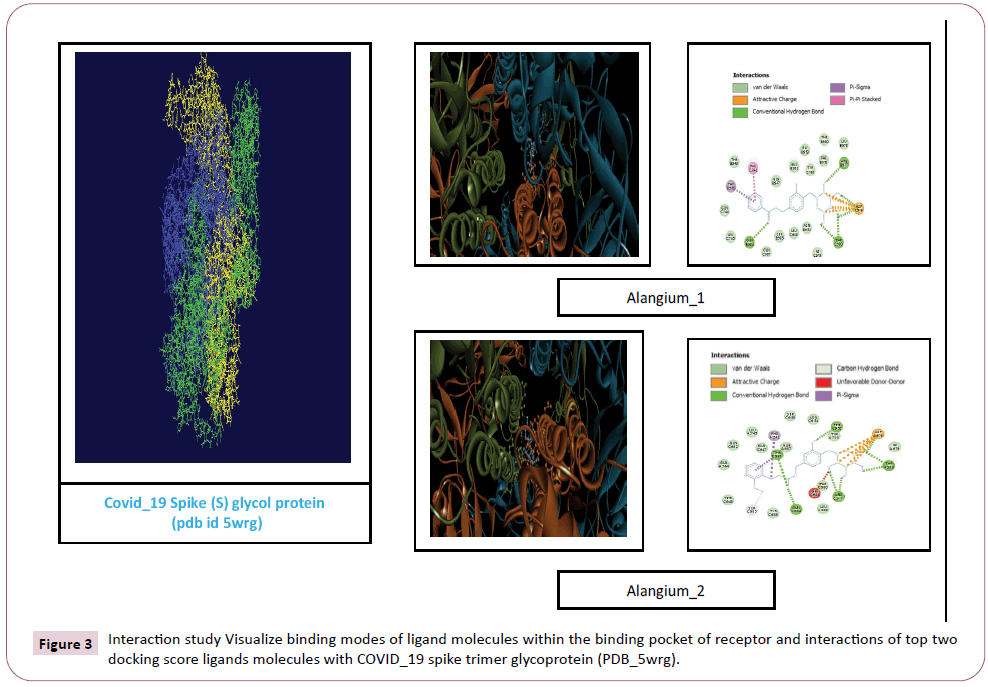
Figure 3: Interaction study Visualize binding modes of ligand molecules within the binding pocket of receptor and interactions of top two docking score ligands molecules with COVID_19 spike trimer glycoprotein (PDB_5wrg).
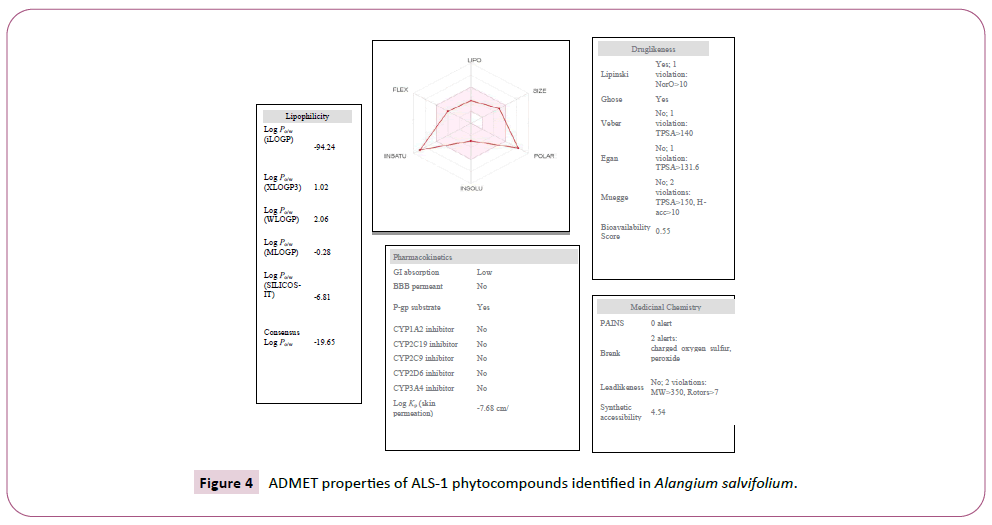
Figure 4: ADMET properties of ALS-1 phytocompounds identified in Alangium salvifolium.
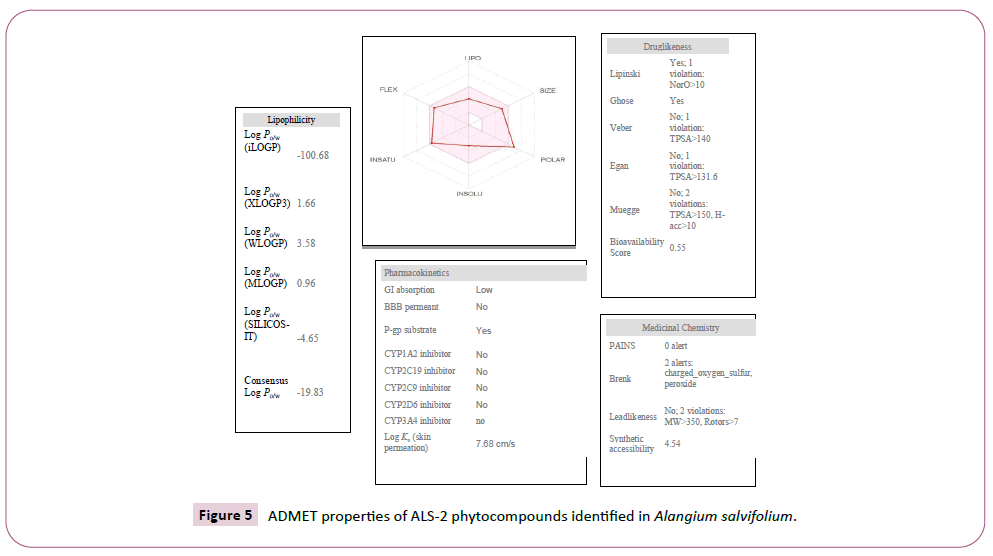
Figure 5: ADMET properties of ALS-2 phytocompounds identified in Alangium salvifolium.
| S. No |
Ligands code |
IUPAC name |
Chemical name |
Molecular weight |
Element involve in docking |
Molar refractivity |
Molecular formula |
Number of heavy atoms |
Num of H bond acceptor |
Num of
bond donar |
Solubility |
| 1 |
ALS-1 |
4 (benzoyloxy)methyl
-2hydroxyphenoxy
tetrahydorxy
hexoxone 1, 2, 3, 4, 5,
pentaium |
Salviifoside-A |
437.32 |
AC HD OA |
81.88 |
C16H21O14 |
28 |
4 |
5 |
Hydrophilic |
| 2 |
ALS-2 |
----- |
Salviifoside-B |
419.35 |
AC HD OA |
94.46 |
C17H23O12 |
29 |
12 |
4 |
Hydrophilic |
Table 1 Different chemical and physiological properties of phyto-derivatives of Alangium salvifolium studied medicinal plants (recognized as a ligands).
| Target Receptor |
Ligands |
Dimension Centre (x=25, y=25, z=25 |
No of pose |
RMSD % Lower |
RMSD % Upper |
Mean binding energy |
Virus Spike protein trimer (Mushroom like) structure
(PDBID-5wrg) |
ALS- 1 |
X=190.072, y=190.525
Z=167.9877 |
8 |
55.21 |
44.62 |
-11.6 |
| ALS-2 |
8 |
63.44% |
39.27% |
-10.8 |
Table 2 Mean values of docking energies (kcal/mol) and standard deviation for each skeletal type of Alangium salvifolium phyto-derivatives as ligands with virus spike (S) protein targets.
| Target Receptor |
Ligands |
Number of pose |
Number of chain involve in pocket |
Number of amino acid involve in active site ( Bond formation)
(chain name and residue number) |
Virus Spike Protein (Trimer)
Assembly |
ALS-1 |
8 |
2 |
C-ASP (976), C-THR (980), C-ILE (979), B-ASN (951), C-LEU (983), B-SER (985), C-GLN (987), B-GLN (984), C-LEU (745), C-GLN (744), B-THR (988).B-THR943), C-PHE (741)B-GLN (947), B-GLY (981, B-GLY (953), C-TYR (738), B-PHE (952), B-THR (980), B-LEU (978), B-ARG (977) |
| ALS-2 |
8 |
2 |
A-ASP (976), A-ILE (979), A-THR (980), C-ARG (977), C-THR (980), C-GLY (981), C-ARG (977), A-LEU (983), C-GLN (984), C-TYR (989), C-SER (985), C-THR (943), A-GLN (992), A-LEU (745), C-PHE (741), C-THR (988), A-GLN (987), C-SER (950), C-ASN (951), C-PHE (952), A-THR (738) |
Table 3 The binding interactions analysis of Virus Spike (Receptor) protein with Alangium salvifolium phyto-derivatives (ligands).
Conclusion
Hence, we conclude that the Alangium salvifolium phytoderivatives may have suitable potential, neither inhibits the virus fusion nor blocks the massive conformation changes by interaction between HR-1 and fusion peptide junction. This study suggests that the chosen phytoderivatives will be further optimized and fine investigated and evaluated for active pharmaceutical ingredients (API) or whenever drug goes under lab and run phases, we may be able to use as Alangium salvifolium crude extract to prevent and treat COVID-19 infection.
Conflicts of Interest
The authors have declared that no conflict of interest exists
28956
References
- https://www.who.int/news-room/commentaries/detail/criteria-for-releasing-covid-19-patients-from-isolation
- Huang C, Wang Y, Li X, Ren L, Zhao J, et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497-506.
- Jiang S, Du L, Shi Z (2020) An emerging coronavirus causing pneumonia outbreak in Wuhan, China: calling for developing therapeutic and prophylactic strategies. Emerg Microbes Infect 9: 275 -277 (2020).
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270-273.
- Liu DX, Inglis SC (1991) Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology 185: 911-917.
- Martinon F, Chen X, Lee AH, Glimcher LH (2010) TLR activation of the transcription factor XBP1regulates innate immune responses in macrophages. Nat Immunol 11: 411-418.
- Fung S, Liu DX (2019) Human coronavirus: Host-pathogen interaction. Annu Rev Microbiol 73:529-557.
- Matthews K, Schäfer A, Pham A, Frieman M (2014) The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol J 11: 209-213.
- Liu S, Xiao G, Chen Y, He Y, Niu J, et al. (2004) Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 363: 938-947.
- Lu L, Liu Q, Zhu Y, Chan KH, Qin L, et al. (2014) Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun 5: 3067.
- i F, Li W, Farzan M, Harrison SC (2005) Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309: 1864-1868.
- Venkateshwarlu R (2011) Phytochemistry and pharmacology of Alangium salvifolium: A review.J of Pharmacy Res 4: 1423-1425.
- Shravya S, Vinod BV, Sunil C (2017) Pharmacological and phytochemical studies of Alangium salvifolium Wang. - A review. Bulletin of Facts of Pharmacy 55: 217-222.
- Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Methods Mol Biol 1263: 243-250.
- https://www.nigms.nih.gov/education/fact-sheets/Pages/structural-biology.aspx
- Houghton PJ, Ren Y, Howes MJ (2006) Acetylcholinesterase inhibitors from plants and fungi. Nat Prod Rep 23: 181-199.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev 46: 3-26.
- Seeliger D, De Groot BL (2010) Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 24: 417-422.
- Alexey L, Dmitrii F, Vladimir P (2000) PASS: prediction of activity spectra for biologically active substances. Bioinformatics 16: 747-758.
- Yuan Y, Cao D, Zhang Y, Ma J, Qi J, et al. (2017) Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8: 15092.
- Xia S, Yan L, Xu W, Agrawal AS, Algaissi A, et al. (2019) A pan-coronavirus fusion inhibitor targeting the the HR1 domain of human coronavirus spike. Sci Adv 5: eaav4580.
- Xia S, Zhu Y, Liu M, Lan Q, Xu W, et al. (2019) Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol 17: 765-767.

