Palabras clave: Enfermedad Creutzfeldt- Jakob; sangre; priones; hemofilia.
Key words: Creutzfeldt-Jakob`s Disease; blood; prions; haemophilia.
Hemoterapia: Pasado, presente y enfermedades emergentes
Desde hace más de 20 años, la hemoterapia se ha basado en lograr la máxima seguridad de las transfusiones de sangre total, de sus componentes o de los derivados plasmáticos ya que la disponibilidad de una sangre segura y fiable constituye un factor crucial para el progreso de la medicina y el mantenimiento de la Salud Pública. Esta inquietud nacía en aquel entonces cuando, aproximadamente, 1 de cada 100 unidades de sangre transmitía el virus de la inmunodeficiencia humana (VIH) o el virus de la hepatitis C (VHC).
A pesar de las mejoras introducidas desde aquella primera transfusión de Denys [1], en cuanto a la selección de los donantes y en cuanto a las innovaciones tecnológicas, tanto serológicas como de detección de agentes infecciosos mediante las técnicas de amplificación de ácidos nucleicos, debemos permanecer todavía muy atentos para prevenir futuras y potenciales catástrofes iatrogénicas, porque no en vano se calcula, por ejemplo, que 1 de cada 2000 transfusiones de plaquetas podrían contener agentes infectivos. El paludismo o el dengue se están convirtiendo en un nuevo problema de Salud Pública que afecta también a la seguridad transfusional. Hace no muchos años estas infecciones se asociaban a los viajes exóticos y esporádicos y que se solucionaban manteniendo en cuarentena durante unos meses a los afectados que pudieran ser posibles donantes de sangre. La situación actual es muy diferente. Miles de inmigrantes y emigrantes se trasladan en pocas horas, cada vez más y de forma exponencial, por todo el mundo y el Trypanosoma cruzi puede llegar en el 50% de los inmigrantes que proceden de zonas endémicas para este agente. Pero quien dice Trypanosoma puede decir Babesia, Leishmania, Virus del Nilo, Virus de la Gripe Aviar, Síndrome Respiratorio Agudo o Virus Chikungunya. En otras palabras, las enfermedades emergentes y reemergentes que condicionan, extraordinariamente, la seguridad transfusional [2].
Porque la realidad, como bien define Zessin [3], es que estamos ante la “livestock revolution” o lo que es lo mismo “revolución del ganado” y la globalización, que se caracterizan por un aumento del libre intercambio de animales y de productos alimenticios. Las condiciones de la vida moderna son una consecuencia de la globalización que determina en muchos de los casos la prevalencia de ciertos factores responsables de las llamadas enfermedades emergentes. Así, se puede hablar de ciertos cambios ecológicos tales como aquellos debidos a la agricultura o al desarrollo económico o fruto de anormalidades climáticas, cambios demográficos humanos o de comportamiento, viajes, comercio, tecnología e industria, adaptación y cambios en los microorganismos, y la falta, en general, de medidas relacionadas con la Salud Pública. En lo que respecta a los agentes patógenos una característica llamativa de las enfermedades emergentes y reemergentes es el aumento del rango de diversidad que ahora existe desde las bacterias, rickettsias, hongos, protozoos, helmintos, hasta las proteínas (priones) pasando por los virus.
De esta forma, la perspectiva epidemiológica no permite una predicción y prevención a largo plazo respecto a la mayor parte de los nuevos patógenos, y más bien sólo ofrece una estimación teórica y grosera de los riesgos para una determinada población de patógenos. La correlación mínima entre la producción animal, las enfermedades de origen animal y las enfermedades humanas exige reconsiderar conceptos, métodos y estructuras, estableciendo nuevas medidas para la coordinación de situaciones de emergencia contra las enfermedades que traspasan las barreras ínter especie.
Encefalopatías Espongiformes Transmisibles
La Encefalopatía Espongiforme Bovina (EEB) ha puesto de actualidad a un conjunto de enfermedades que afectan a los animales y al hombre. A lo largo del tiempo estas enfermedades han recibido diferentes denominaciones, algunas poco rigurosas desde un punto de vista académico, que aludían al presumible agente causal, todavía hoy insuficientemente conocido. Así, se habla de enfermedades causadas por virus lentos, por viroides, viriones o virus no convencionales y, más recientemente, de enfermedades por priones [4,5].
Hasta hace 30 años la más conocida, la Enfermedad de Creutzfeldt-Jakob (ECJ), era una forma rara de demencia desconocida para muchos facultativos. Ahora el nombre nos es familiar, y ello ha sido debido a la aparición en 1996, de la Encefalopatía Espongiforme Bovina (EEB) más conocida como Enfermedad de las “vacas locas”. La posible asociación de la nueva variante en humanos de la Enfermedad de Creutzfeldt-Jakob (vECJ) con la EEB convierte a esta rara enfermedad en centro de atención de autoridades sanitarias, de profesionales de la asistencia médica y de la población en general. Además, el hecho de que la vECJ pueda suponer un riesgo en transfusiones de sangre y sus derivados, ha causado cierta intranquilidad social.
Las Encefalopatías Espongiformes Transmisibles (EET) tienen como causa un agente infeccioso si bien también se puede presentar como enfermedad genética o esporádica. Su denominación se basa en dos de sus principales características: la espongiosis cerebral y la transmisibilidad. En humanos, pueden presentarse la Enfermedad de Creutzfeldt-Jakob (ECJ) en sus formas esporádica, iatrogénica y familiar; el Kurú; la Encefalopatía de Gerstmann-Sträussler-Scheinke (GSS); el Insomnio Familiar Letal (esporádica o familiar) (FFI/SFI) y la variante de la Enfermedad de Creutzfeldt-Jakob (vECJ) que es exclusiva de humanos.
Enfermedad de Creutzfeldt-Jakob
La Enfermedad de Creutzfeldt-Jakob (ECJ) afecta al cerebro produciendo la desestructuración total neuronal haciendo que el cerebro presente una apariencia de esponja y pierda toda su funcionalidad nerviosa [6]. Se trata de una enfermedad de naturaleza degenerativa y de pronóstico fatal cuya prevalencia es, aproximadamente, de una persona por millón a nivel global. En España, en el año 2006, se registraron 30 casos de ECJ. Esta enfermedad puede darse de forma esporádica (idiopática), hereditaria o adquirida. El 80-90% de los casos en el mundo son del tipo esporádico. La ECJ es un mal neurológico transmisible secundario a implantes de duramadre, córnea o tratamientos con hormona de crecimiento de origen hipofisario, fundamentalmente. El agente causal es una proteína llamada prión (PrP). Si bien los casos hereditarios e infecciosos están perfectamente documentados, la causa de la aparición del prión se desconoce en la mayor parte de los casos informados. Los síntomas son los típicamente neurológicos que afectan al movimiento, así como somnolencia extrema y una máxima torpeza. Los grados más graves de la enfermedad pueden cursar con síntomas psiquiátricos como puede ser un estado severo de demencia. Aunque se han investigado mucho, durante los últimos años, los aspectos bioquímicos y de replicación de los priones, mucho queda todavía por saber sobre los mecanismos moleculares que dan lugar a la neurodegeneración del sistema nervioso, aunque es muy posible que se encuentren alterados los mecanismos de biogénesis celular de estas proteínas que controlan el “tráfico” intracelular, el plegamiento o el direccionamiento extracelular [7]. También se encuentra en estudio la reciente hipótesis de que la infección por priones pueda requerir el contacto íntimo entre un determinado tejido, como pudiera ser el muscular, y neuronas o terminaciones nerviosas infectadas [8]. Tampoco se sabe si una pequeña cantidad de prión puede ser determinante para padecer la enfermedad, aunque se puede intuir, como para cualquier otro tipo de infección, que su desencadenamiento pueda ser dosis dependiente o dependiente de otros cofactores [9].
Hasta el momento actual, se han descrito tres causas para la ECJ: i) esporádica o espontánea que es la más habitual, representando el 85% de todos los casos; ii) familiar o hereditaria en la que interviene un componente genético que se trata de una mutación puntual en el gen que codifica la proteína del prión y que hace que ésta se pliegue defectuosamente, precipite y desestructure el cerebro, y iii) iatrogénica que se produce cuando se adquieren los priones bien por la dieta en carne de animales afectados o de sus derivados, por transplantes o bien por la administración de sangre o derivados que contienen priones y que proceden de personas que ya padecen la enfermedad o que se encuentran asintomáticas. La variante de la Enfermedad de Creutzfeldt- Jakob (vECJ) se trata de la forma en humanos del “mal de las vacas locas”.
El agente causal es una proteína infecciosa denominada prión que no se inactiva por los métodos antivirales habituales [10]. Estos nuevos agentes infecciosos se transmiten a través de la sangre ya que se encuentran adheridos a la membrana externa de los linfocitos. Además, son difícilmente filtrables, es decir, no se retienen, debido a su muy pequeño tamaño, en los filtros convencionales de esterilización. Por otra parte, ya que son proteínas que, con su estructura normal, se encuentran en el organismo de forma fisiológica y no patológica, no se reconocen como extrañas por el organismo ya que conservan las regiones antigénicas intactas, por lo que no se genera una respuesta inmunológica eficaz de rechazo. Esta enfermedad presenta un periodo ventana o asintomático que puede ser muy largo, de hasta 40 años, teniendo en cuenta que niños que recibieron la hormona del crecimiento en los años 60´ y que se obtenía de la hipófisis de cadáveres, ahora están desarrollando la vECJ. De este hecho se deriva una problemática difícilmente controlable relacionada con la calidad de las donaciones ya que además, hoy por hoy, no está disponible una prueba “in vitro” para realizar en sangre y detectar los priones, pudiéndose tan solo realizar el diagnóstico por biopsias de tejido nervioso o por biopsia “postmorten”.
Es fácil imaginar, por tanto, la situación de un donante de sangre que se encuentre en el periodo asintomático de la enfermedad y que lo desconozca. Esto unido a la imposibilidad de realizar una prueba diagnóstica sistemática a los donantes, hace de éstas personas, donantes de alto riesgo desde el punto de vista de Salud Pública. Además, tampoco, hoy por hoy, existe un tratamiento para la curación de la enfermedad y el desenlace es fatal. Tan solo se ha descrito una prolongación del tiempo de incubación de la enfermedad, al reducir la acumulación del prión en el cerebro, administrando pentosal polisulfato por vía intraventricular [11].
El prión (PrP) en su forma normal (PrPc), cuya función se desconoce, se localiza en el cerebro y en otras regiones del organismo en el hombre y en muchos animales. La proteína anormal del prión o PrPsc, es químicamente idéntica a la forma normal pero su conformación espacial es diferente, haciéndola resistente a los procesos de degradación normal en la célula e induciéndose su acumulación en varios tejidos, especialmente en el sistema nervioso central donde se produce el daño más severo [12]. Además, los priones se transmiten a través de la sangre [13]. El progreso de la enfermedad se caracteriza por una pérdida de tejido neuronal que da lugar a un aspecto del cerebro de esponja característico. Un hecho importante es que no hay respuesta perceptible por parte del sistema inmune contra el prión anómalo, aún teniendo una conformación espacial diferente. Por otra parte, la proteína anormal del prión es resistente a la mayor parte de los métodos usados para inactivar bacterias y virus. Como consecuencia de esto, los priones no se inactivan totalmente por el calor, la luz ultravioleta u otros procedimientos estándares de esterilización, tales como el hipoclorito sódico en las concentraciones habituales. El autoclavado simple (un solo ciclo) no sirve para desnaturalizar la proteína anormal del prión presente en los instrumentos quirúrgicos.
La proteína anormal inicial del prión se puede producir espontáneamente (explicación posible para la ECJ esporádica); estar ligada a una anormalidad genética heredada del gen PrP (ECJ familiar); o adquirirse a través de material biológico de personas infectadas o por productos derivados del plasma (ECJ iatrogénica). Es de destacar que una gran proporción de los pacientes con ECJ esporádica y la vECJ presentan el polimorfismo del codón 129 (metionina/metionina) que se trata de un factor genético condicionante [14]. Este genotipo, que está presente en el 40% de la población general, probablemente predispone a la conversión del prión normal en el anormal que se asocia a la enfermedad.
En los casos en que se desarrolla la enfermedad se produce un cambio conformacional del prión lo que favorece el acumulo de la proteína priónica en las células neuronales, hecho que altera su función y desemboca, finalmente, en la vacuolización del tejido cerebral.
Variante de la Enfermedad de Creutzfeldt-Jakob (vECJ)
El primer caso de EEB en humanos surge en 1994, una década después de su aparición en las vacas. En 1996 se describe, por primera vez, una nueva enfermedad, relacionada con la EEB, que se define como variante de la ECJ (vECJ) cuyo origen era o podía ser el consumo de carne de vacas infectadas por la EEB sobre la base de la coincidencia espacio-temporal y en contra de la idea de que las EET animales no afectaban al hombre. En los últimos años numerosas pruebas experimentales han demostrado que la vECJ es la manifestación en humanos de la EEB a través de lo que ya se denomina “salto de las barreras inter-especie”.
El número de casos de EEB, alcanza su máximo (cerca de 200000 vacas) en 1992, y disminuye hasta una casuística mínima en 1998, dejando una cascada epidemiológica de la vECJ, en humanos, debido al consumo de productos bovinos contaminados, que no se restringió al Reino Unido ya que han surgido casos en otros países ya sea como resultado de la importación de animales enfermos o de suplementos dietéticos como es el caso de las harinas cárnicas.
Parece tratarse de una enfermedad con características clínicas y anatomopatológicas propias y claramente diferenciales respecto a la ECJ clásica o esporádica. Aparece en adultos más jóvenes con un desarrollo más lento, de unos 14 meses frente a los 4 de la ECJ clásica. La ECJ clásica se presenta a nivel mundial con una incidencia de 1 caso por millón de habitantes y por año, mientras que la vECJ se restringe, por ahora, casi en la totalidad de los casos, al Reino Unido. La ECJ clásica se manifiesta con un cuadro de demencia o ataxia de progresión rápida, mientras que la vECJ cursa como enfermedad psiquiátrica, con o sin síntomas sensitivos, de evolución lenta hasta el estadio neurológico grave. Las fases finales de ambas enfermedades son muy similares [15].
Desde la aparición del primer caso de la vECJ, se han referido ya 162 casos en el Reino Unido, 21 en Francia, 4 en Irlanda, 1 en Italia, 3 en Estados Unidos, 1 en Canadá, 1 en Arabia Saudita, 1 en Japón, 2 en Holanda, 1 en Portugal y 1 en España [16].
Factores plasmáticos de la coagulación y la variante de la Enfermedad de Creutzfeldt-Jakob en el tratamiento de las hemofilias
En la década de los años 70´ la hemofilia se comenzó a tratar con concentrados de factores VIII o IX que se obtenían mediante el fraccionamiento de plasma humano. Después, hacia los años 90´, comenzaron a comercializarse los factores recombinantes —los mal llamados factores sintéticos— que se obtienen, en un laboratorio de Ingeniería Genética y Biología Molecular, a partir de determinadas células de mamífero, convenientemente modificadas con los genes correspondientes de Factor VIII o Factor IX.
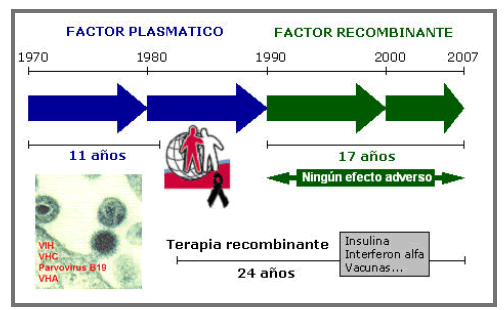
La historia de los factores plasmáticos y recombinantes es claramente diferente desde el punto de vista de la seguridad farmacológica. Así, los factores plasmáticos, tan solo a los 11 años desde el inicio de su utilización, ya habían desencadenado la gran pandemia iatrogénica de la infección por el virus de la inmunodeficiencia humana (VIH) y, poco más tarde, en los años 90, por el antes mal llamado virus de la hepatitis no A, no B y que ahora conocemos como el virus de la hepatitis C (VHC). Ambas infecciones han causado miles de muertes en la población hemofílica diezmando en más de la mitad el número de pacientes hemofílicos en todo el mundo. Todavía, estos dos tipos de virus, a pesar de encontrarse inactivados en los concentrados plasmáticos, causarán muchas más muertes a corto y largo plazo, debido a las resistencias que se producen frente a los fármacos antirretrovirales, debido a los efectos adversos acumulativos de estos fármacos (fundamentalmente fallos renales, pancreáticos o de infarto de miocardio por aumento del colesterol en pacientes relativamente jóvenes), y por la derivación a cirrosis hepática y hepatocarcinomas en ese 60% de pacientes que no responden a los tratamientos actuales.
En contrapartida a esta trágica historia de los productos plasmáticos, se encuentran los factores recombinantes que ofrecieron la esperanza y la tranquilidad en el tratamiento de la hemofilia, pero sobre todo seguridad que se ha consolidado sobre la base de no haberse producido todavía ningún acontecimiento adverso infectivo o de otro tipo, ni leve ni grave, en sus casi ya 17 años de aplicación en la clínica hemofílica.
La historia de los factores plasmáticos se pensó que podía cambiar con la aplicación, desde finales de los años 80´, de los métodos de inactivación viral por calor y después, a finales de los 90´, con los métodos de doble inactivación a altas temperaturas y con solventes-detergentes, pero esto no fue así. Nuevas amenazas han surgido y surgirán con las llamadas enfermedades emergentes, fomentadas por el extraordinario movimiento demográfico del hombre, que son capaces de atravesar, incluso, las barreras interespecie de animales a hombre. Esto ha hecho que los factores plasmáticos, que ya eran y son muy seguros frente a virus con cubierta lipídica como el VIH o el VHC, se vuelvan de nuevo vulnerables a nuevos agentes infecciosos como puede ser la Enfermedad de Creutzfeldt-Jakob.
Entre los años 1997 y 2000 los Servicios Nacionales de Bancos de Sangre y la Agencia de Protección de la Salud del Reino Unido [17], notificaron la existencia de donantes de sangre que habían desarrollado la vECJ con posterioridad a la donación de sangre.
Desde el inicio de la aparición de la vECJ, en el Reino Unido, se especuló con el riesgo teórico de la transmisión por la sangre y los hemoderivados. Aunque la evidencia epidemiológica no sugería que la forma esporádica se pudiera transmitir por la vía sanguínea, sí se reconoció, en cambio, la posible transmisión por esta vía de la nueva variante de la ECJ debido a sus características, en comparación a la forma clásica, especialmente en lo que se refiere a la implicación del sistema linforreticular. Esta hipótesis se demostraba después, en diciembre de 2003, cuando se confirmaba el contagio en un paciente por una transfusión de concentrados de hematíes; después fue un segundo caso en julio de 2004, un tercero en 2006 y el más reciente en enero de 2007.
En el caso de España se desató la intranquilidad cuando en 2005 se anunciaba el primer caso de la vECJ en una mujer. Este caso hubiera pasado inadvertido si no se hubiera dado la circunstancia de que aquella mujer, fallecida por esa enfermedad, había sido donante de sangre en España durante varios años. Su sangre se utilizó para el fraccionamiento de plasma y se obtuvieron entre otros fármacos diversas inmunoglobulinas, concentrados de plaquetas y de hematíes, así como concentrados de Factor VIII plasmático para el tratamiento de la hemofilia A. La compañía farmacéutica española de fraccionamiento que preparó este factor no dio la señal de alarma o si la dio aseguró que no existía riesgo de contagio ni de padecer la vECJ, así que todos los lotes que se prepararon de Factor VIII, a partir del plasma contaminado por priones, se administraron íntegramente a pacientes hemofílicos. El resultado, más de 300 hemofílicos, de nueve Comunidades Autónomas, utilizaron el producto.
Riesgo de infección y de desarrollo de la variante de la Enfermedad de Creutzfeldt-Jakob a través de la sangre y de los productos derivados del plasma
Se ha demostrado experimentalmente, en modelos animales, que los priones se encuentran presentes en la sangre y que se transmiten por esta vía [12,13]. Además, las células blancas —que se pueden encontrar en los concentrados de hematíes y otros componentes sanguíneos para uso transfusional— presentan una infectividad igual que el plasma [18]. Por esta razón, existe un riesgo de contagio a través de los instrumentos quirúrgicos, a través de un trasplante, por transfusión de sangre o por el tratamiento con productos derivados de un plasma contaminado.
Según Farrugia [19], los parámetros que afectan al riesgo son, esencialmente, los siguientes: i) número de donaciones; ii) porcentaje de donantes de sangre infectados por la vECJ; iii) volumen de plasma por donación; iv) número de unidades infectivas de la vECJ por mL de plasma; v) número de unidades de producto elaboradas en el proceso de producción; vi) reducción logarítmica del número de unidades infectivas durante el proceso de producción, y vii) cantidad de producto que ha utilizado el paciente. En síntesis, el posible bajo riesgo de infección de dichos concentrados se debe parcialmente a dos factores, de una parte, a la dilución de una donación contaminada con miles de donaciones “limpias” en el mismo lote de plasma, y de otra, al propio proceso de fabricación, si incluye pasos que eliminen el agente infeccioso tales como la leucodepleción, la precipitación selectiva, la nanofiltración y la cromatografía de intercambio iónico.
No se pueden ofrecer datos que cuantifiquen el riesgo de infección a través de los productos plasmáticos ya que se desconocen muchas cuestiones sobre los agentes patógenos que producen la enfermedad. Aún así, sí se puede hacer una estimación del riesgo, entendida ésta como una valoración sobre la base de modelos teóricos epidemiológicos muy complejos. Esta estimación será, por tanto, tan solo, una orientación para tomar precauciones especiales a efectos de Salud Pública y para evitar cualquier transmisión posible de la vECJ, o para llevar a cabo un seguimiento clínico y racional de los pacientes que han sido receptores de productos contaminados con priones.
Se considera riesgo probable cuando se han recibido productos derivados de un plasma de donantes que desarrollaron posteriormente la vECJ. En otoño de 2003 se llevó a cabo por la consultora “Det Norske Veritas” (DNV) [20], una estimación del riesgo de infección por priones cuando se utilizan factores plasmáticos, datos de riesgo que se han aceptado por el Comité Consultivo de las Encefalopatías Espongiformes (SEAC), por el Comité para la Seguridad Microbiológica de la sangre y de tejidos, y por el Comité para la Seguridad de los Medicamentos. Este cálculo de riesgo es el que se acepta todavía hoy como único dato de referencia [19].
En la actualidad a pesar de los eficaces métodos de inactivación que se aplican a los factores plasmáticos, no se puede decir que estos productos sean totalmente seguros ya que siempre se preparan a partir de miles de donantes de sangre y, en estas circunstancias, las enfermedades emergentes no son controlables. Hoy día los factores plasmáticos sólo son seguros frente a aquellos virus que presentan cubierta lipídica ya que éstos son sensibles a los métodos que utilizan solventedetergente y calor. No sucede lo mismo con aquellos virus que no presentan esta cubierta, como es el virus de la hepatitis A (VHA) o el parvovirus B19, u otros emergentes de este tipo que puedan surgir. Por supuesto los métodos de inactivación viral actuales no sirven para los priones que son resistentes a todos estos métodos. Estas circunstancias no se dan, por el contrario, con los factores de origen recombinante
Aunque la estimación del riesgo varía en función de los pasos de aclaramiento de priones durante el fraccionamiento del plasma y dependiendo de los distintos autores y las distintas formas de preparación de los hemoderivados [21-24], los concentrados de Factor VIII y de Factor IX que se obtienen a partir de los crioprecipitados y criosobrenadantes respectivamente, se considera que presentan un riesgo de medio a alto respecto a la vECJ si es que el plasma hubiera estado contaminado por priones. Con respecto a la albúmina que se utiliza como estabilizante de los concentrados recombinantes de Factor VIII, se considera de riesgo bajo.
El riesgo es bajo si se tienen en cuenta todas aquellas fases de aclaramiento, o de disminución del riesgo, que se pueden dar durante el proceso de preparación de los concentrados y que atenúan los niveles de prión. Aún así se debe ser cauteloso y tomar precauciones adicionales para proteger mejor a la población hemofílica. Lo que sí se sabe es que el tiempo o periodo asintomático antes de que aparezcan los primeros síntomas de la enfermedad puede ser muy largo, de hasta 10 o 20 años, y también se sabe, ya que se ha demostrado en modelos animales, que la vECJ sí se transmite realmente a través de la sangre. Se deberán, por tanto, tomar medidas alternativas mediante tratamientos que no impliquen la utilización de productos derivados del plasma, véanse los productos recombinantes, al menos, hasta que se conozca el mecanismo íntimo de contagio —niveles de prión, vías de transmisión— y se establezca la fiabilidad de los métodos de aclaramiento de priones. Será determinante conseguir una prueba de detección de los priones en la sangre para descartar a los donantes de sangre portadores de la vECJ. Lo que sí está claro, sin ser ninguna novedad en Medicina, es que existe un riesgo y que en ningún caso se podrá pensar que es cero o inexistente. Aún así, hoy por hoy, no hay ningún caso descrito de la vECJ en aquellos pacientes hemofílicos que han recibido concentrados obtenidos a partir de un plasma que contenía priones, aunque en Medicina la ausencia de evidencia no significa evidencia de ausencia [25]. No se debe descartar tampoco, que algunos casos hayan pasado inadvertidos por el desconocimiento actual sobre la enfermedad y por no disponerse de un método adecuado de diagnóstico.
En cuanto a las posibilidades de desarrollar la vECJ, después de una infección por priones, decir algo es todavía más comprometedor ya que no se dispone de datos estadísticos suficientes para arriesgar una predicción, ni de bases moleculares para establecer un mecanismo etiopatogénico
Medidas para la prevención
Las medidas de prevención derivan de la clasificación hecha por la Organización Mundial de la Salud relativa a los grados de infectividad de órganos, tejidos y fluidos a partir de los datos obtenidos en modelos experimentales animales de diferentes especies.
Se deberán tomar, en principio, aquellas medidas de tipo general que se adoptan para cualquier tipo de infección, en este caso las medidas generales sobre la población de no consumir carne o productos derivados de animales infectados. También se adoptarán aquellas propias de toda buena práctica clínica dirigidas a la “bioseguridad” como son la descontaminación del material quirúrgico, en general, y del de odontología en particular, intentando siempre que sea posible, aunque sea más costoso, utilizar material de un solo uso e implantar los métodos más recientes y novedosos de inactivación de priones [26-29].
Por los conocimientos actuales sobre las Encefalopatías Espongiformes, hoy por hoy, no hay evidencia de un riesgo de transmisión por el contacto habitual y próximo a través de la piel, saliva, ni por el acto sexual, aunque sí se recomienda, a efectos de Salud Pública, evitar situaciones de contacto íntimo o de donación de óvulos o espermatozoides en personas contagiadas por priones.
Lo que sí es aconsejable, como medida más conveniente, desde el punto de vista epidemiológico cuando se trata de una enfermedad infecciosa, es adoptar o desarrollar una muy buena Farmacovigilancia, en este caso una Hemovigilancia [30,31], que incluirá la detección, clasificación y análisis de los efectos no deseados de una transfusión sanguínea o de sus derivados con el fin de corregir las causas y prevenir su reincidencia. A este respecto el Parlamento Europeo, en su Directiva 2002/98/CE [32], incluye, en su capítulo V, la obligación, para todos los Estados Miembros, de adoptar todas las medidas necesarias para garantizar la trazabilidad desde el donante al receptor y viceversa así como sobre la notificación de todos los efectos y reacciones adversas graves relacionadas con la extracción, la verificación, el tratamiento, el almacenamiento y la distribución de sangre y de sus componentes sanguíneos que puedan influir en la calidad y seguridad de los mismos. También sobre las reacciones adversas graves que se pudieran producir durante una transfusión, o después de ella, y que puedan atribuirse a la calidad y la seguridad de la sangre o de sus componentes
Será fundamental en este caso el seguimiento tanto de los donantes de sangre, potenciales enfermos asintomáticos de la vECJ, como de los pacientes que hayan recibido sangre contaminada con priones ya sea en forma de sangre entera, plasma o derivados y componentes sanguíneos como pueden ser los concentrados de hematíes o de plaquetas, pero fundamentalmente los concentrados de factores de la coagulación ya que estos productos presentan una muy alta concentración de proteínas entre las que se pueden encontrar los priones
Con relación a la sangre, se deberán considerar muy escrupulosamente, por una parte, el Real Decreto 1088/05 de 16 de septiembre [33] y, de otra, la Orden SCO/322/2007, de 9 de febrero [34], por la que se establecen los requisitos de trazabilidad y de notificación de reacciones y efectos adversos graves de la sangre y de los componentes sanguíneos, así como de las buenas prácticas clínicas [35], en donde se regula la selección de los donantes, la leucodepleción —o eliminación de los linfocitos de la sangre—, el registro de los donantes de sangre y la información a los receptores de sangre o de sus derivados. Se deberán excluir como donantes de riesgo a aquellas personas que hayan residido más de 1 año acumulativo en el Reino Unido —Inglaterra, Escocia, Gales, Irlanda del Norte, Isla de Man e isla del Canal— durante el periodo comprendido entre 1980 y 1996, ambos inclusive. Se deberá aplicar la leucodepleción universal [36] de la sangre ya que está claramente demostrado que los niveles de priones son muy elevados en el sistema linforreticular [37], e instar para aplicar otros métodos de eliminación de priones en los hemoderivados como son varios ciclos de calentamiento, la nanofiltración y la cromatografía de intercambio iónico [38].
En el marco del Sistema de Vigilancia Epidemiológica de las Encefalopatías Espongiformes Humanas se valorarán de forma periódica los casos notificados al Registro Español de la Enfermedad de Creutzfeldt-Jakob y de aquellos casos con antecedentes de donación de sangre o sus componentes. Los receptores de sangre, plasma o derivados contaminados serán informados. Además, se deberá establecer un sistema de coordinación con el Registro de Encefalopatías del Instituto de Salud Carlos III, con relación a los receptores de componentes sanguíneos y hemoderivados contaminados. Se deberá, por último, promover el uso apropiado de la sangre y tejidos y tomar medidas alternativas, cuando ello sea posible, como puede ser el uso de concentrados recombinantes para el tratamiento de las hemofilias.
Con relación a los pacientes que hayan recibido derivados de sangre de un donante que presentaba la vECJ, se les debe ofrecer la posibilidad de ser informados, previo consentimiento privado, y aconsejarlos para no donar sangre, ni tejidos, ni órganos ya que se consideran, a partir de ese momento, personas de riesgo desde el punto de vista de Salud Pública. En este sentido el Ministerio de Sanidad y Consumo Español ha estado coordinando, recientemente, una campaña informativa en España a través de las distintas Comunidades Autónomas y sus Consejerías de Sanidad junto a las asociaciones de pacientes de coagulopatías congénitas y los hematólogos responsables que conforman la Comisión Científica de la Real Fundación “Victoria Eugenia” de Hemofilia.

Presentes y futuras alternativas para la máxima prevención
Es de sobra conocido que respecto a la sangre hay tres axiomas fundamentales: el primero, es que la sangre siempre será un bien escaso que necesitará de donación; el segundo, que el riesgo cero no existe para el que recibe sangre y, el tercero, que la sangre al ser un producto biológico, nunca estará libre totalmente de contaminación por virus, bacterias o priones, conocidos en la actualidad, o por otros agentes todavía por descubrir. En este estado de cosas, ya que se va a seguir precisando sangre, un objetivo inmediato es intentar administrar el llamado “oro rojo” en los hospitales de la forma más segura y adecuada [39]. Además, también hay que decir que, aunque las enfermedades emergentes representan una preocupación real y muy actual, la donación de sangre es un hecho inevitable vista la poca oferta de donaciones y la gran demanda que existe de sangre y de sus hemoderivados [40].
Estas razones, tan simples y a la vez tan complejas, llevan a adoptar una serie de medidas para hacer de la sangre y de sus derivados algo seguro. En primer lugar, las medidas encaminadas a la selección de los donantes en función de factores epidemiológicos y demográficos respecto a determinadas infecciones y en función de aquellas pruebas serológícas negativas respeto a distintas infecciones que se pueden detectar en sangre. En segundo lugar, la utilización de fármacos que evitan el plasma como fuente de obtención, como son los productos y medicinas recombinantes que, después de casi 25 años sin producir efectos secundarios, se han erigido como una alternativa terapéutica muy eficaz y segura [41]. En este sentido, muchos países están llevando a cabo políticas sanitarias encaminadas a la incentivación y recomendación del uso de productos recombinantes siempre y cuando no estén contraindicados y así lo permitan los factores socio-económicos. Concretamente, en España, ya se ha redactado una Proposición no de Ley del Ministerio de Sanidad y Consumo para la implantación progresiva de productos recombinantes para el tratamiento de las coagulopatías congénitas [42].
Por su parte, se han establecido consensos internacionales sobre las distintas alternativas a la transfusión de sangre alogénica [43]. La administración de Factor VII recombinante activado, especialmente útil para reducir el sangrado y/o los requerimientos transfusionales en diversos procedimientos médicos o quirúrgicos que cursan con hemorragia masiva no controlada con los métodos convencionales; la aprotinina como antifibrinolítico, dosis dependiente, que inhibe la tripsina, la plasmina y la calicreína plasmática y tisular; la desmopresina que incrementa la adhesión plaquetaria —por aumento de la expresión del receptor de la GPIb plaquetaria— y los valores plasmáticos de los factores VIII y de von Willebrand desde sus lugares de producción en la célula endotelial del sinusoide hepático; la eritropoyetina recombinante que estimula la eritropoyesis al inhibir la apoptosis de los precursores eritroides, y que promueve la proliferación y maduración a eritrocitos; los perfluorocarbonados de reemplazo del volumen intravascular con capacidad para fijar gases como el O2; y la recuperación perioperatoria de sangre autóloga para ser devuelta al paciente en forma de concentrados de hematíes en suero salino, son ejemplos reales de estas alternativas. Además, las empresas farmacéuticas libran una batalla contra reloj para encontrar un sustituto de la sangre —“la sangre artificial”— mediante procedimientos biotecnológicos. Las llamadas células rojas “enmascaradas” y los compuestos de fluorocarbono, son ejemplos de los futuros sustitutivos de las células rojas transportadoras de oxígeno a los tejidos. Por su parte, los fragmentos de plaquetas conjugados con albúmina sérica constituyen la posibilidad del futuro para prescindir de la perfusión de plaquetas en casos de trombocitopenias [44]. Más recientemente, las polihemoglobinas conjugadas a enzimas antioxidantes, recubiertas de membranas lipídicas, abren una nueva perspectiva para el futuro [45].
Conclusión final
La historia de la Medicina ha demostrado, sin lugar a dudas, que el riesgo cero no existe en cualquier práctica médica y que, por tanto, se debe adoptar el mayor número de precauciones posibles en relación a la Salud Pública, aun cuando las estadísticas, como sucedió en 1979 respecto al SIDA y, como puede suceder ahora para la vECJ, indiquen que existe un mínimo riesgo. Hoy, para que sirva como ejemplo, el VIH ha infectado a unos 65 millones de personas y el SIDA se ha cobrado más de 25 millones de vidas [46].
1184
References
- Myhre BA. The first recorded blood transfusions: 1656 to 1668. Transfusion 1990; 30: 358-62.
- Lejarazu RO. Los animales como vectores de las enfermedades emergentes. Med Clin (Barc) 2005; 124: 16-8.
- Zessin KH. Emerging diseases: A global and biological perspective. J Vet Med B Infect Dis Vet Public Health 2006; 53(Suppl 1): 7-10.
- Watts JC, Balachandran A, Westaway D. The expanding universe of prion diseases. PLoS Pathog 2006; 2: 152-63. [consultado 15-03-2007] Disponible en: https://pathogens.plosjournals.org/archive/1553-7374/2/3/pdf/10.1371_journal.ppat.00200 26-S.pdf.
- Fornai F, Ferrucci M, Gesi M, Bandettini di Poggio A, Giorgi FS, Biagioni F, et al. A hypothesis on prion disorders: are infectious, inherited, and sporadic causes so distinct? Brain Res Bull 2006; 69: 95- 100.
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–44.
- Tatzelt J, Schatzl HM. Molecular basis of cerebral neurodegeneration in prion diseases. FEBS J 2007; 274: 606-11.
- Dlakic WM, Grigg E, Bessen RA. Prion infection of muscle cells in vitro. J Virol 2007. (En prensa) [consultado 15-03-2007] Disponible en: https://jvi.asm.org/cgi/content/abstract/JVI .02628-06v1
- Fasano C, Campana V, Zurzolo C. Prions: protein only or something more? Overview of potential prion cofactors. J Mol Neurosci 2006; 29: 195-214.
- Sorgato MC, Bertoli A. Physiopathologic implications of the structural and functional domains of the prion protein. Ital J Biochem 2006; 55: 222-31.
- Parry A, Baker I, Stacey R, Wimalaratna S. Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulphate. J Neurol Neurosurg Psychiatry 2007. [consultado 15-03-2007] Disponible en: https://jnnp.bmj.com/cgi/rapidpdf/jnnp.200 6.104505v1. 12. Peden AH, Ritchie DL, Head MW, Ironside JW. Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am J Pathol 2006; 168: 927-35.
- Ludlam CA, Turner ML. Managing the risk of transmission of variant Creutzfeldt Jakob disease by blood products. Br J Haematol 2006; 132: 13-24.
- Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, et al. Predicting susceptibility and incubation time of human-to-human transmission of vECJ. Lancet Neurol 2006; 5: 393-8.
- Will RG, Ward HJ. Clinical features of variant Creutzfeldt-Jakob disease. Curr Top Microbiol Immunol 2004; 284: 121-32.
- Grupo europeo y de países colaboradores para el estudio de la vECJ (The European and Allied CountriesCollaborative Study Group of CJD). [consultado 15-03-2007]: Disponible en: https://www.eurocjd.ed.ac.uk/EUROINDEX. htm (Section 3; results; table 4).
- Agencia de Protección de la Salud del Reino Unido (Health Protection Agency). [consultado 15-03-2007]: Disponible en: https://www.hpa.org.uk/infections/topics_a z/cjd/menu.htm.
- Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004; 363: 417-21.
- Farrugia A, Ironside JW, Giangrande P. Variant Creutzfeldt-Jakob disease transmission by plasma products: Assessing and communicating risk in an era of scientific uncertainty. Vox Sang
- Consultora DNV para la evaluación del riesgo por la vECJ (Det Norske Veritas). [consultado 15-03-2007]: Disponible en: https://www.dnv.com/consulting/news/risko finfectionfromvariantcjdinblood.asp.
- Brown P, Rohwer RG, Dunstan BC, MacAuley C, Gajdusek DC, Drohan WN. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 1998; 38: 810-6.
- Brown P, Cervenakova L, McShane LM, Barber P, Rubenstein R, Drohan WN. Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an
- explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion 1999; 1169-78. 23. Foster PR. Prions and blood products. Ann Med 2000; 32: 501-13.
- Foster PR, Welch AG, McLean C, Griffin BD, Hardy JC, Bartley A, et al. Studies on the removal of abnormal prion protein by processes used in the manufacture of human plasma products. Vox Sang 2000; 78: 86-95.
- Cerezo C, Cordón F, Solanas P. Enfermedad de las “vacas locas”: La ausencia de evidencia no significa evidencia de ausencia. Aten Primaria 2001; 28: 188-92.
- Almazán J, Alvarez-Quiñones M, Avellanal F, Bermejo F, Calero M, Cuadrado N, et al. Enfermedad de Creutzfeldt-Jakob y otras Encefalopatías Espongiformes Transmisibles Humanas. Guía de información y recomendaciones para personal sanitario. Consejo Interterritorial del Sistema Nacional de Salud 2003. Ministerio de Sanidad y Consumo.
- Avellanal F, Almazán J, Calero M, Mahillo I, Martínez P, De Pedro J, et al. Encefalopatias espongiformes transmisibles humanas y atención odontológica. Centro Nacional de Epidemiología 2006. Ministerio de Sanidad y Consumo.
- Vadrot C, Darbord JC. Quantitative evaluation of prion inactivation comparing steam sterilization and chemical sterilants: proposed method for test standardization. J Hosp Infect 2006; 64: 143-8.
- Solassol J, Pastore M, Crozet C, Perrier V, Lehmann S. A novel copper-hydrogen peroxide formulation for prion decontamination. J Infect Dis 2006; 194: 865-9.
- Verdú JJ, Verdú J, Soler S. Hemovigilancia activa: un sistema que evalúa los efectos adversospostransfusionales no detectados por el sistema de hemovigilancia convencional. Med Clin (Barc) 2006; 127: 156.
- Lozano M, García-Villaescusa R. Hemovigilancia: Más allá de la transfusión. Haematologica (Ed esp) 2003; 88: 188- 202.
- Directiva 2002/98/CE del Parlamento Europeo y del Consejo de 27 de enero de 2003 por la que se establecen normas de calidad y de seguridad para la extracción, verificación, tratamiento, almacenamiento y distribución de sangre humana y sus componentes y por la que se modifica la Directiva 2001/83/CE. [consultado 15-03- 2007]: Disponible en: https://www.cgcom.org/internacional/europ a_dia/2003/pdf/98_directiva_2002_98_seg uridad_sangre.pdf.
- Real Decreto 1088/2005, de 16 de septiembre, por el que se establecen los requisitos técnicos y condiciones mínimas de la hemodonación y de los centros y servicios de transfusión. [consultado 15- 03-2007]: Disponible en: https://www.boe.es/boe/dias/2005/09/20/p dfs/A31288-31304.pdf.
- Orden SCO/322/2007, de 9 de febrero, por la que se establecen los requisitos de trazabilidad y de notificación de reacciones y efectos adversos graves de la sangre y de los componentes sanguíneos. [consultado 15-03-2007]: Disponible en: https://www.boe.es/boe/dias/2007/02/17/p dfs/A07010-07016.pdf.
- Ortiz P, Mingo A, Lozano M, Vesga MA, Grifols JR, Castrillo A, et al. Guide for transfusion of blood components. Med Clin (Barc) 2005; 125: 389-96.
- Gregori L, McCombie N, Palmer D, Birch P, Sowemimo-Coker SO, Giulivi A, et al. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet 2004; 364: 529-31.
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, et al. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol 2004; 203: 733-9.
- Burnouf T, Padilla A. Current strategies to prevent transmission of prions by human plasma derivatives. Transfus Clin Biol 2006; 13: 320-8.
- Mayordomo J. Cómo administrar el “oro rojo” de los hospitales mientras llega la sangre artificial. Reportaje en EL PAIS. [consultado 15-03-2007]: Disponible en: https://www.elpais.com/articulo/salud/administrar/oro/rojo/hospitales/elpsalpor/20050 510elpepisal_1/Tes.
- Seitz R, von Auer F, Blumel J, Burger R, Buschmann A, Dietz K, et al. Impact of vCJD on blood supply. Biologicals 2007. (En prensa) [consultado 15-03-2007] Disponible en: https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=17320412&query_hl=11&itool=pubmed_docsum. 41. Liras A. Terapia recombinante: ¿Del escepticismo a la aplicación de elección?. Editorial invitado. Rev Act Farmacol Terp 2005; 3: 165-7. [consultado 15-03-2007]Disponible en: https://www.socesfar.com/pdf/aft3.pdf.
- Proposición no de Ley presentada por el Grupo Parlamentario Socialista del Congreso para promover un estudio para la posible aplicación del factor recombinante como alternativa terapéutica en la
- hemofilia. Boletín Oficial de las Cortes Generales de España, 18 de diciembre de 2006. Serie D, General, núm. 482.
- Leal R, Alberca I, Asuero S, Bóveda JL, Carpio N, Contreras E, et al. Documento “Sevilla” de Consenso sobre Alternativas a la Transfusión de Sangre Alogénica. Med Clin (Barc) 2006; 127(Suppl 1): 3-20. [consultado 15-03-2007]: Disponible en: https://www.aehh.org/img/marcela01.pdf?a ehh=8629fd5dadf47b671552b57a3399bf3 4.
- Kim HW, Greenburg AG. Toward 21st century blood component replacement therapeutics: artificial oxygen carriers, platelet substitutes, recombinant clotting factors, and others. Artif Cells Blood Substit Immobil Biotechnol 2006; 34: 537- 50. [consultado 15-03-2007] Disponible en: https://taylorandfrancis.metapress.com/con tent/u032127rv00m2068/.
- Chang TM. Evolution of artificial cells using nanobiotechnology of hemoglobin based RBC blood substitute as an example. Artif Cells Blood Substit Immobil Biotechnol 2006; 34: 551-66. [consultado 15-03- 2007] Disponible en:https://taylorandfrancis.metapress.com/con tent/x4213678267j2578/.
- Organización de las Naciones Unidas para el SIDA. [consultado 15-03-2007]: Disponible en: https://data.unaids.org/pub/GlobalReport/2 006/200605-FS_globalfactsfigures_es.pdf.

