Introducción
La leucodistrofia metacromática (LM) es una enfermedad lisosomal del grupo de las esfingolipidosis producida por la deficiencia de la enzima arilsulfatasa A (EC 3.1.6.8). Los bajos niveles de actividad enzimática afectan el metabolismo del cerebrósido sulfato provocando la acumulación intralisosomal del sulfátido esfingolípido 3'-O-sulfogalactosilceramida [1] en la sustancia blanca del sistema nervioso central y periférico; particularmente en las vainas de mielina que rodean las células nerviosas, así como de lactosil sulfátido en los riñones, vesícula biliar, vejiga y otros órganos [2]. La incidencia de esta enfermedad es de 1:40 000 y el patrón de herencia asociado es autosómico recesivo. El gen codificante para la enzima se ha localizado en el cromosoma 22 región q13-31. También se han reportado, aunque en menor número, casos producidos por la deficiencia de la proteína saponina B la cual actúa como activador de la arilsulfatasa A en la ruptura de los esfingolípidos. El gen que codifica para la misma se encuentra en el cromosoma 10 [3,4].
El cuadro clínico se caracteriza por nerviosismo, hipersensibilidad, convulsiones, atrofia del nervio óptico, demencia, parálisis y ceguera. Las diferentes formas de evolución se relacionan con la mutación existente. De acuerdo con la edad de comienzo de los síntomas se reportan las siguientes formas: infantil tardía, juvenil, juvenil tardía y adulta.
La forma infantil tardía, es la más frecuente y debuta alrededor del primer y segundo año de vida con la adopción de una locomoción atáxica y afectaciones de tipo mental y muscular como la hipotonía. Se produce afectación del sistema nervioso periférico, atrofia del nervio óptico y un proceso que culmina en la muerte aproximadamente cuatro o cinco años después del comienzo de los síntomas y signos clínicos. Desde el punto de vista bioquímico, los niveles de actividad enzimática en estos casos son muy bajos o totalmente ausentes.
La forma juvenil aparece entre los 4 y 6 años de vida, produciéndose cambios en el comportamiento, con pérdida de las funciones mentales que repercuten en el desarrollo intelectual y por tanto en las actividades escolares. Aún cuando el curso es más lento aparece la epilepsia, la ataxia y una regresión motora de forma general, culminando en la muerte al llegar a la edad de 20 años. En estos pacientes los niveles de actividad de la enzima son bajos aunque sin llegar a los de la forma infantil.
En el caso de la forma juvenil tardía y adulta, la enfermedad comienza entre los 6 y 16 años y después de los 16, respectivamente. La evolución de la enfermedad es lenta con predominio de problemas del comportamiento que pueden influir en la escuela o centro laboral, convulsiones y afectaciones en el control muscular que implican trastornos motores. También se desarrollan con frecuencia trastornos psiquiátricos de progresión lenta. La arilsulfatasa A muestra una actividad residual [2,5,6].
Es importante conocer que sujetos normales pueden tener niveles extremadamente bajos de arilsulfatasa A, complicando la detección de portadores entre los familiares de enfermos de LM, los casos presintomáticos y el diagnóstico prenatal.
Han sido pocos los casos que se han reportado de esta enfermedad en la etapa neonatal. La sintomatología puede caracterizarse por retardo, detención o involución del desarrollo psicomotor, irritabilidad, dificultad en la alimentación y síndrome piramidal con adopción de postura en opistótonos. Es frecuente la aparición de ceguera por atrofia del nervio óptico [4].
En el presente trabajo se realiza un reporte del primer caso de leucodistrofia metacromática en el período neonatal diagnosticado clínica y bioquímicamente en nuestro país.
Caso clínico
Paciente de 7 días de nacida, femenina, de piel blanca, hija de padres no consanguíneos de 31 años el padre y 24 la madre. No se refieren antecedentes patológicos familiares para ambos padres y el resto de los familiares. Como antecedente prenatal se presenta dilatación pielocalicial unilateral ligera seguida por ultrasonido. Parto a las 41,5 semanas, peso y talla al nacer de 3600g y 51 cm respectivamente.
Diagnóstico clínico
Cerca de la semana de nacida la paciente comenzó a presentar succión pobre e hipotonía. Los padres acudieron al servicio de neonatología del Hospital Pediátrico William Soler (La Habana, Cuba), donde se ingresó para estudio y tratamiento. En su evolución posterior comenzó a presentar opistótonos. Se realizó punción lumbar y Tomografía Axial Computarizada (TAC) de cráneo.
Diagnóstico bioquímico
Inicialmente se realizaron como pruebas bioquímicas cualitativas el ensayo de la Ninhidrina [7] y el del Benedict [8]. Seguidamente se determinó el perfil de azúcares acumulados en orina, así como el perfil correspondiente a los aminoácidos elevados en suero y orina mediante una cromatografía en placa delgada [9]. Como método confirmatorio se determinó el nivel de actividad de la enzima arilsulfatasa A en homogeneizado de leucocitos; utilizando como control negativo del ensayo la muestra de un individuo supuestamente sano [10,11].
Discusión
Las leucodistrofias son enfermedades que se clasifican como desmielinizantes y provocan afectación en el sistema nervioso central producto de un defecto enzimático de carácter hereditario. En este grupo se encuentran esfingolipidosis como la LM y la enfermedad de Krabbe, la X-adrenoleucodistrofia, una enfermedad peroxisomal y la enfermedad de Canavan, entre otras. Todas ellas comparten la sintomatología con algunas pequeñas diferencias y están estrechamente relacionadas con la edad de aparición.
Teniendo en cuenta la diversidad y origen de estas enfermedades se hace necesario trazar una línea de trabajo que incluya pruebas en orina, suero y análisis neurofisiológicos que permitan discriminar entre ellas, llegando así a un diagnóstico certero. Los exámenes como el TAC son de gran importancia ya que guían el diagnóstico hacia la determinación de una deficiencia enzimática en particular [4,5].
En el caso de esta paciente se realizó una punción lumbar, arrojando resultados negativos. El TAC de cráneo al ser un examen neurológico, permite discriminar entre las leucodistrofias, mostrando en este caso una leucoatrofia.
Los casos de LM en la etapa neonatal no son los más ampliamente reportados; sin embargo se han reportado dos casos, cuyo diagnóstico estuvo basado en estudios histológicos. Uno de ellos, corresponde a un varón que fallece a las seis semanas de nacido presentando períodos de apnea, cianosis, debilidad general, movimientos tónicos-clónicos, y en tejidos provenientes de la materia blanca se encontró depósito de material granular metacromático [6]. El otro caso, fue una hembra que desarrolló desde el nacimiento cianosis, apnea y debilidad generalizada y muere a las 20 horas de nacida [12].
Esto muestra que en aquellos casos donde se sospeche una LM, los hallazgos clínicos deben corroborarse mediante pruebas bioquímicas en período inmediato a la sospecha para lograr corroborar el diagnóstico presuntivo.
El ensayo de Benedict confirmó la presencia de azúcares reductores como la glucosa, galactosa, fructosa, lactosa, ranosa y xilulosa, así como de otras sustancias reductoras como el ácido sílico y el ácido homogentísico [8]. La aparición de un color rojo ladrillo intenso en la orina de la paciente mostró la acumulación de alguno de ellos. En la cromatografía en placa delgada se observó en el perfil correspondiente a la muestra de la paciente una banda a la altura de los glúsidos galactosa y/o glucosa, al ser comparado con el patrón compuesto por una mezcla de los azúcares lactosa, galactosa, glucosa, fructosa y xilosa (figura 1).
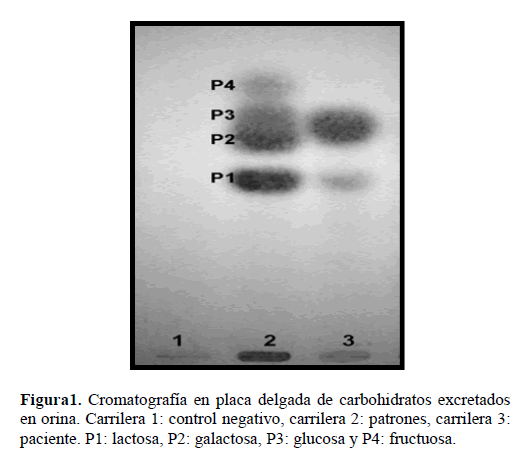
Figura 1. Cromatografía en placa delgada de carbohidratos excretados en orina. Carrilera 1: control negativo, carrilera 2: patrones, carrilera 3: paciente. P1: lactosa, P2: galactosa, P3: glucosa y P4: fructuosa.
El ensayo de la ninhidrina evidenció la presencia de aminoácidos en orina al colorearse la misma de un color púrpura intenso. Este resultado se corroboró mediante una cromatografía en placa delgada al observarse bandas a nivel de los aminoácidos leucina y/o isoleucina, metionina y/o valina y arginina en la muestra de orina, así como bandas a nivel de los aminoácidos leucina y/o isoleucina en la muestra de suero (figura 2).

Figura 2. Cromatografía en placa delgada de aminoácidos presentes en suero y excretados en orina. Carrilera 1: paciente (orina), carrilera 2: paciente (suero), carrilera 3: patrón A, carrilera 4: patrón B, carrilera 5: patrón C, carrilera 6: control negativo. A1: cistina, A2: lisina, A3: aspártico, A4: alanina, A5: valina, A6: fenilalanina; B1: cistina, B2: arginina, B3: treonina, B4: prolina, B5: triptófano, B6: leucina; C1: histidina, C2: glicina, C3: glutámico, C4: tirosina, C5: metionina, C6: isoleucina.
Los hallazgos clínicos y los resultados cualitativos positivos se confirmaron mediante la determinación cuantitativa de la actividad de la enzima. El ensayo para la enzima arilsulfatasa A se realizó en un homogeneizado de leucocitos; utilizando la muestra de un individuo supuestamente sano como control negativo. Los valores obtenidos fueron de 51.75 nmol/mg/h para el paciente y de 202.5 nmol/mg/h para el control, siendo la actividad enzimática de la paciente de un 25 % con respecto al control. A partir de estos resultados se pudo establecer definitivamente el diagnóstico de leucodistrofia metacromática por deficiencia de la enzima arilsulfatasa A.
Aún cuando no existe un tratamiento para esta enfermedad, la posibilidad de realizar un diagnóstico certero y definitivo permite el asesoramiento genético a la familia y a la pareja en particular. Es importante una estrecha relación y una correcta metodología de trabajo entre las especialidades participantes, dígase neonatólogos, genetistas, radiólogos, bioquímicos, farmacéuticos y analistas de laboratorio en general.
1096
References
- Gieselmann V, Franken S, Klein D, Mansson JE, Sandhoff R, Rauch RL et al. Metachromatic leukodystrophy: consequences of sulphatide accumulation. Acta Paediatrica. 2003; 92(s443):74-9.
- Kolodny EH, Fluharty AL. and multiple sulfatase deficiency: sulfatide lipidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D Eds. The metabolic and molecular basis of inherited disease, 7th edn. New York: McGraw Hill, 1995: 2693–739.
- Fluharty AL. Arylsulfatase A Deficiency. [en línea] Seattle: University of Washington, 2006. [consulta: 28 enero 2008].
- Terradas-Covisa JML. Diagnóstico y tratamiento de las Leucodistrofias. [en línea] Madrid: Servicio de Neurologia. Hospital Niño Jesús, 1998. [consulta: 28 enero 2008].
- Moore T, Steiner RD. Metachromatic leukodystrophy [en línea] Los Angeles: University of California at Los Angeles Medical Center, 2006. [consulta: 28 enero 2008].
- Feigin I. Diffuse cerebral sclerosis (metachromatic leukoencephalopathy). Am J Pathol 1954; 30: 715–37.
- Slocum RH, Cummings JG. Aminoacids analysis of physiological samples. In Hommes FA, ed. Techniques in diagnostic human biochemical genetics: a laboratory manual. New York: Wiley-Liss Inc; 1991:87-126.
- Shih VE, Mandell R and Sheinhait I. General metabolic screening tests. In Hommes FA, ed. Techniques in diagnostic human biochemical genetics: a laboratory manual. New York: Wiley-Liss Inc; 1991:45-68.
- Shih VE, Lai N, Mandell R and Hommes FA. Monosaccharides and disaccharides. In Hommes FA, ed.Techniques in diagnostic human biochemical genetics: a laboratory manual. New York: Wiley-Liss Inc; 1991:69-75.
- Galjaard H. Genetic metabolic diseases. Early diagnosis and prenatal analysis. Amsterdam: Elsevier/North-Holland Biomedical Press; 1980:215-36, 823.
- Wenger DA, Williams C. Screening for lysosomal disorders In Hommes FA, ed. Techniques in diagnostic human biochemical genetics: a laboratory manual. New York: Wiley-Liss Inc; 1991:587-617.
- Bubis JJ, Adlesberg L. Congenital metachromatic leukodystrophy. Report of a case. Acta Neuropathol 1966; 6:298.

