Keywords
Mitochondria; Cerebral ischemia; Antioxidants; Reactive oxygen species
Introduction
Cerebral ischemia limits the delivery of substrates, mainly oxygen, glucose and impairs the energy requirement to the brain [1]. It is a leading cause of death in industrialized and developing countries. There is growing evidence that mitochondria play a major role in both necrotic and apoptotic neuronal cell death after cerebral ischemia [2,3]. Cerebral ischemia induced mitochondrial swelling, opening of mitochondrial permeability transition pore leading to either necrotic or apoptotic cell death, is currently being explored intensely [4-6]. Existing concepts advocate that mitochondrial swelling may be the result of membrane permeability transition initiated by a variety of stimuli. The stimuli for membrane permeability shift differ between ischemia alone versus ischemia with reperfusion as a consequence of the generation of reactive oxygen species. Mitochondrial dysfunction and oxidative stress are mutually dependent and reinforce damages that play a central role, not only in brain aging, but also in neurodegenerative disease [7]. Overproduction of reactive oxygen species (ROS), which may arise either from mitochondrial electron-transport chain or excessive stimulation of NAD(P)H, results in oxidative stress, a toxic process that can play a crucial role in damage of cellular components, including lipids and membranes, proteins, and DNA. At moderate levels, ROS participate in physiological signaling by contributing to the adjustment of brain function to cellular metabolism and metabolic supply. Mitochondrial dysfunction acts through a number of destructive pathways, including excessive production of ROS resulting in oxidative modification of mitochondrial proteins. This in turn causes impairment of oxidative phosphorylation, contributing to the onset and progression of disease. 10-(6-plastoquinonel) decyltriphenyl-phosphonium (SkQ1), mitoquinone (MitoQ), Coenzyme Q10 (CoQ10) and Methylene blue (MB) are antioxidants that selectively target mitochondria and protect it from oxidative damage and which have been shown to decrease mitochondrial damage in animal models of oxidative stress [8,9].
Effects of mitochondrial dysfunction on ischemic brain
Mitochondria play an essential role in the life and death of living cells, performing several fundamental regulatory processes. Destruction of the mitochondrial energy metabolism is the immediate cause of mitochondrial dysfunction and disruption of oxidative phosphorylation a key mechanism of producing adenosine triphosphate (ATP) in cerebral ischemia [10,11]. The maintenance of the mitochondrial membrane potential (MMP), which helps to establish a proton gradient across the inner mitochondrial membrane to activate the adenosine triphosphate (ATP) synthase to generate high-energy phosphates, is disturbed during cerebral ischemia. Loss of MMP may be a common feature of ischemic destructive processes; these processes favour the progression and initiation of the apoptotic cell death [12,13]. Moreover, mitochondria are main targets and source of oxidative stress, and an excess of ROS has been implicated in the pathogenesis of cerebral ischemia. These oxygen free radicals are main contributors to necrotic or delayed neuronal death and powerful initiators of inflammation and apoptosis [14].
Oxidative stress
Oxidative stress is a phenomenon in which there is an imbalance between free radicals and antioxidants in the living system, which plays a major role in the pathophysiology of neurodegenerative disorder [15]. The brain is at higher risk to the damage caused by oxidative stress due to high content of polyunsaturated fatty acid, high consumption of oxygen, elevated metabolic activity and relatively limited ability to combat with oxidative stress [16]. ROS act as secondary messengers in many intracellular signaling pathways and as mediators of oxidative damage and inflammation [17]. Free radicals can attack directly polyunsaturated fatty acids in membranes and initiate lipid peroxidation (LPO). These features may make the brain a target tissue for the onset and pathogenesis of a number of neurological disorders via oxygen radical production [18,19].
Inflammation
Cerebral ischemia is accompanied by a marked inflammatory reaction that is initiated by ischemia induced expression of cytokines, adhesion molecules, and other inflammatory mediators. Many inflammatory mediators and pro-inflammatory cytokines like TNF-α and IL-1β, are up-regulated during this period. The release of IL-1β is totally dependent on the response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) that attach and bind to pattern recognition receptors (PRRs) to up-regulate proinflammatory gene expression [20,21]. PAMPs are patterns carried by pathogens, such as bacterial endotoxin and DAMPs are commonly endogenous substances released by necrosis.
A number of inflammasome forming PRRs have been identified, including the Nod-like receptors (NLR) family, pyrin domain containing proteins (NLRP1, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4) and AIM2. Among all inflammasomes identified until now, the best characterized and most strongly related with inflammation, is formed by NLRP3 [22]. NLRP3 can be activated by a various groups of disease associated molecules, where it oligomerizes with the adaptor ASC (apoptosis-associated specklike protein containing a caspase recruitment domain) and caspase-1 to form the NLRP3-in?ammasome, resulting in the processing of pro to mature IL-1β and its subsequent release. Massive amounts of ATP are rapidly released into the extracellular space after traumatic or ischemic injuries [23-26] and a high concentration of ATP activates NLRP3 inflammasome [23,27-28]. This indicates that NLRP3 inflammasome senses mitochondrial dysfunction and may explain the frequent association of mitochondrial dysfunction with inflammation. In a renal ischemic model, NLRP3 activation was found to be mediated at least partly via mitochondrial ATP released from disrupted cells [29].
Apoptosis
Mitochondria also plays important role in neuronal apoptosis, primarily the release of proapoptotic factors into the cytoplasm after the opening of mitochondrial transition pore. The first proapoptotic proteins, cytochrome c are released from the membrane space into cytosol [30]. Once released, these proteins activate the caspase dependent mitochondrial pathway and activate Apaf-1, as well as procaspase-9. Together with ATP these proteins form the apoptosome [31]. The aggregation of procaspase-9 leads to the activation of caspase-9 which is most probably an initiator of cytochrome c dependent caspase cascade, leading to the activativation of caspase-3. Caspase-3 has been reported as a key mediator of apoptosis in the ischemic brain, which cleaves many substrate proteins, including poly (ADP-ribose) polymerase (PARP) [32,33] and translocates AIF to the nucleus [34] as well as facilitating the accessibility of nuclear chromatin to endonucleases [35]. Activation of PARP after cleavage by caspase-3 leads to DNA damage. Subsequently, tumour suppressor transcription factor, p53, stops the cell cycle and prompts programmed cell death by up-regulation of pro-apoptotic protein expression, and down regulation of antiapoptotic protein leading to subsequently cell death [36].
Mitochondrial targeted antioxidant
Mitochondria are the primary source of intracellular reactive oxygen species (ROS) and are mainly susceptible to oxidative stress. Oxidative damage to mitochondria has been shown to impair mitochondrial function and lead to cell death via apoptosis and necrosis. There is evidence that antioxidant treatments can ameliorate or delay disease progression in animal models of neurodegenerative diseases. Naturally occurring antioxidants SKQ1, CoQ10, MitoQ and MB have shown protective effects in the ischemic brain. The nature of these antioxidants and their protective role in ischemic brain are explained below and a schematic diagram has been shown in Figure 1.
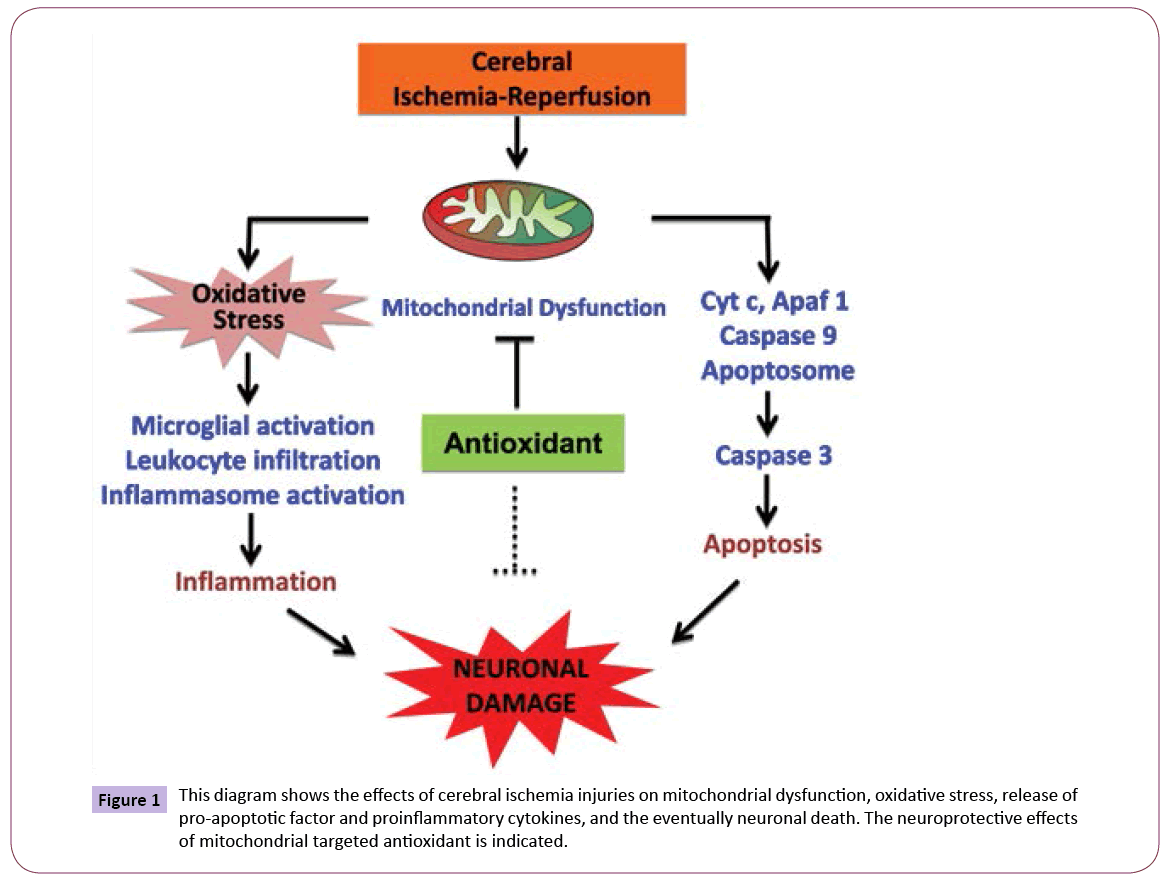
Figure 1: This diagram shows the effects of cerebral ischemia injuries on mitochondrial dysfunction, oxidative stress, release of pro-apoptotic factor and proinflammatory cytokines, and the eventually neuronal death. The neuroprotective effects of mitochondrial targeted antioxidant is indicated.
SKQ1
Mitochondria are important in the aging process, and one of the ways in which they interact with surrounding cell biology by ROS. The lipophilic cation SkQ1 is a mitochondrial targeted antioxidant, which accumulates inside the mitochondria, as these organelles form the only negatively charged compartment in the cell. High membrane potential of the inner mitochondrial membrane together with membrane potential (inside negative) of the plasma membrane and the strong hydrophobicity of SkQ1 results in the accumulation of SkQ1 in the inner leaflet of the inner mitochondrial membrane [37]. Oxidized SkQ1 formed after the scavenging of ROS is rapidly reduced by the mitochondrial respiratory chain, resulting in regeneration of reduced form of SkQ1. It has been observed that SkQ1 diminished the mitochondrial fragmentation (fission) after cerebral ischemia and prevent progression of the apoptotic cascade [38].
CoQ10
CoQ10 is fat soluble quinine with 10 five-carbon isoprenoid units, acts as an anti oxidant, protecting the cells against oxidative damage. CoQ10 is a component of the mitochondrial electron transport chain, transfering electron in the respiratory chain and playing a role in membrane stabilization [39]. CoQ10 is normally involved in a series of enzymatically catalyzed sequential reactions necessary to carry out oxidative phosphorylation via the electron transport chain (ETC). It collects reducing electrons from flavoprotein. Oxidative phosphorylation is a fundamental process used in the production of energy to maintain the brain function through the production of ATP [40,41]. CoQ10 has been reported to decrease brain lactate levels and lessen the diameter of ischemic lesions in animal models [42,43]. CoQ10 improves neurological outcomes and prevents neuronal damage by reducing the production of free radicals and preventing lipid peroxidation, a major cause of damage by free oxygen radicals. In addition, transient cerebral ischemia leads to decrease in tissue levels of CoQ10 [44]. Moreover, it has also been reported that CoQ10 lessens the diameter of ischemic lesions in animal models [45-47].
Mito Q
Mitochondria are vulnerable to oxidative damage and are also a major source of superoxide free radicals. Consequently, mitochondria accumulate oxidative damage that could contribute to mitochondrial dysfunction and cell death in cerebral ischemia. MitoQ is a lipophilic triphenylphosphonium (TPP) cation which helps it to pass easily through the phospholipid bilayer of mitochondria [48,49]. Within mitochondria, MitoQ is reduced by the respiratory chain reaction to its active form ubiquinol which is an effective antioxidant that prevents lipid peroxidation and mitochondrial damage [50]. MitoQ has been tested in a number of animal models of disease: supplementation of rats with MitoQ decreased heart dysfunction, cell death and mitochondrial damage subsequent to ischemia reperfusion in isolated heart [51,52]. It also protected endothelial cell function and damage to mitochondrial enzymes in a rat model of oxidative stress [53]. It has been found that MitoQ prevents ischemia reperfusion induced injury after the release of cytochrome c and caspase activation leading to massive tissue damage and cell death [54].
Methylene blue
Cerebral ischemia is a main cause of mortality and disability worldwide. The treatment of cerebral ischemia is controlled by a therapeutic window consisting of a few hours, and efficient drugs for the treatment of ischemia are extremely limited. MB has been proved to perform various biological functions and has been used for various medical applications [55]. MB crosses the blood–brain barrier and accumulates in the mitochondrial matrix [56,57]. MB has auto-oxidizing property and acts as an electron cycler which allows MB to pass on electrons to the mitochondrial electron transport chain (in the absence of oxygen), thereby enhancing ATP production, cytochrome c oxidase activity and helping in cell survival via bypassing complex I–III activity to generate ATP [58,59]. MB reduces reactive oxygen species production from the mitochondrial electron transport chain species [60,61] which has the potential to minimize ischemic and reperfusion injury.
Mito PBN
Alpha-phenyl-tert-butyl-nitrone (PBN) is a potent free radical scavenger believed to have protective actions in ischemiareperfusion injury of brain by forming adducts of oxygen free radicals including OH radical [62,63]. It significantly reduces the cerebral infarction, neurological deficit, inducible cytochrome (cyclooxygenase-2) levels and activity, inducible nitric oxide synthase, inhibits mechanisms involved in nuclear factor-kB transduction, induces heme oxygenase-1, promotes mitochondrial function, enhances cholinergic function via acetyl cholinesterase inhibition, and inhibits calcium channels in ischemic brain [64-67].
Conclusion and Future Direction
Mitochondrial targeted antioxidants are one of the most important therapies for providing neuroprotection in cerebral ischemia. A large body of evidence supports the essential role of mitochondrial oxidative stress in ischemic brain injuries and despite the unsatisfactory results of non-targeted antioxidants in clinical trials, there is promising evidence in favor of beneficial effects of mitochondrial targeted antioxidants in ischemic brain injuries. These antioxidants SKQ1, CoQ10, MitoQ and MB reduce the mitochondrial oxidative stress either by direct antioxidant or indirectly through protection of mitochondrial structure and its function. Experimental studies will be needed to establish the significant impact of these treatments in both animal and clinical models of cerebral stroke.
It may be possible to combine these antioxidants with other therapeutic agents, to extend the therapeutic window of the drugs or of the effects of the mitochondrial targeted antioxidant itself. The use of mitochondrial targeted antioxidant in combination with other therapeutic agents may lead to the re-examination of the many neuroprotective drugs that failed at the clinical level, as many drugs may not have been studied in the most effective possible manner. However, it is clear that more research is still needed to understand their biological significance as well as how they can be effectively applied in additional clinical conditions.
Acknowledgement
This research was supported by Research Grant (NS086929) from the National Institutes of Neurological Disorders and Stroke, National Institutes of Health.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this review.
6763
References
- Watts LT, Lloyd R, Garling RJ, Duong T (2013) Stroke Neuroprotection: Targeting Mitochondria.Brain Sci3:540-560.
- Liu J, Li J, Yang Yi, Wang X, Zhang Z, et al. (2014) Neuronal apoptosis in cerebral ischemia/reperfusion area following electrical stimulation of fastigial nucleus. Neural Regen Res 9:727-34.
- Niizuma K,Yoshioka HChen H,Jung JE, et al. (2010 ) Mitochondrial and apoptotic neuronal death signaling pathways incerebral ischemia.Biochim Biophys Acta 1802:92-9.
- Song T, Liu J, Tao X, Deng JG (2014)Protection effect of atorvastatin in cerebral ischemia-reperfusion injury rats by blocking the mitochondrial permeability transition pore. Genet. Mol. Res 13: 10632-10642
- Li JYu WLi XT,Qi SHLi B (2014) The effects of propofol onmitochondrialdysfunction following focalcerebral ischemia-reperfusion in rats.Neuropharmacology 77: 358-68.
- Hagberg H, Mallard C, Rousset CI, Thornton C (2014) Mitochondria: hub of injury responses in the developing brain. Lancet Neurol 13: 217-32
- Ruszkiewicz J,Albrecht J (2015) Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem Int pii: S0197-0186(15)00004-2.
- Miclescu A, Sharma HS, Martijin C, Wicklund L (2010) Methylene blue protects the cortical blood brain barrier against ischemia-reperfusion- induced disruption. Crit Care Med 38: 2199
- Skulachev MV, Antonenko YN, Anisimov VN, Chernyak BV, Cherepanov DA, et al. (2011) Mitochondrial-targeted plastoquinone derivatives.Effect on senescence and acute age-related pathologies. Curr Drug Targets 12: 800-826.
- Sanderson TH,Reynolds CA,Kumar R,Przyklenk K,Hüttemann M (2013) Molecular mechanisms ofischemia-reperfusion injury inbrain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 47: 9-23.
- Panickar KS , Anderson RA (2011) Effect of Polyphenols on Oxidative Stress and Mitochondrial Dysfunction in Neuronal Death and Brain Edema in Cerebral Ischemia. Int J Mol Sci 12: 8181-8207.
- Wang S,Li Y,Song X,Wang X,Zhao C,et al.(2015) Febuxostat pretreatment attenuates myocardial ischemia/reperfusion injury viamitochondrialapoptosis. J Transl Med 13:209.
- Sun J, Li YZ, Ding YH, Wang J, Geng J,et al. (2014) Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro andcerebral ischemia/reperfusion injury in vivo. BrainRes 1589:126-39.
- Lee JC,Won MH(2014) Neuroprotection of antioxidant enzymes against transient globalcerebral ischemiain gerbils. Anat Cell Biol47: 149-56.
- Tabassum R, Vaibhav K, Shrivastava P, Khan A, Ahmed ME, et al. (2015) Perillyl alcohol improves functional and histological outcomes against ischemia-reperfusion injury by attenuation of oxidative stress and repression of COX-2, NOS-2 and NF-κB in middle cerebral artery occlusion rats. Eur J Pharmacol 747:190-199.
- Panickar KS,Qin B,Anderson RA (2014) Ischemia-induced endothelial cell swelling and mitochondrial dysfunction are attenuated by cinnamtannin D1, green tea extract, and resveratrol in vitro. Nutr Neurosci Apr 28.
- Waldbaum S, Patel M (2010) Mitochondrial dysfunction and oxidative stress a contributing link to acquired epilepsy. Journal of Bioenergetics and Biomembranes 42:449-455.
- Ates S, Cayli E (2007) Neuroprotection by resveratrol against traumatic brain injury in rats. Molecular and Cellular Biochemistry 294:137-144.
- Tsai MJ, Liao JF, Lin DY (2010 Silymarin protects spinal cord and cortical cells against oxidative stress and lipopolysaccharide stimulation. Neurochemistry International 57:867-875.
- Chen GY, Nuñez G (2010) Sterile inflammation sensing and reacting to damage. Nat Rev Immunol 10:826-837.
- Takeuchi O, Akira S (2010) Pattern recognition receptors and in?ammation.Cell 140: 805-820.
- Cassel SL, Sutterwala FS (2010) Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol 40:607-611.
- Khakh BS, North RA (2006) P2X receptors as cell-surface ATP sensors in health and disease. Nature 442: 527-32.
- Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50: 413-92.
- Latini S, Corsi C, Pedata F, Pepeu G (1996) The source of brain adenosine outflow during ischemia and electrical stimulation. Neurochem Int, 28: 113-118.
- Peng W, Cotrina ML, Han X, Yu H, Bekar L, et al. (2009) Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A 106: 12489-12493.
- Gombault A, Baron L, Couillin I (2012) ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol 3: 414.
- Dubyak GR (2012) P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell Microbiol 14: 1697-1706.
- Iyer SS, Pulskens WP, Sadler JJ, B utter LM, Teske GJ, et al. (2009) Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 106: 20388-20393.
- Elmore S (2007) Apoptosis a review of programmed cell death. Toxicol Pathol 35:495-516
- Love S (2003) Apoptosis and brain ischaemia. Prog Neuropsychopharmacol Biol Psychiatry 27:267-282.
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, et al. (1998) Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci18:3659-3668.
- Haile WB,Echeverry R,Wu F,Guzman J,An J,et al. (2010) ecrosis factor-like weak inducer ofapoptosisand fibroblast growth factor-inducible 14 mediatecerebral ischemia-induced poly(ADP-ribose) polymerase-1 activation and neuronal death. Neuroscience 171:1256-1264.
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, et al. (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297:259-263
- Duriez PJ, Shah GM (1997) Cleavage of poly (ADP-ribose)polymerase: a sensitive parameter to study cell death. Biochem Cell Biol 75:337-349
- Culmsee C, Mattson MP (2005) p53 in neuronal apoptosis. Biochem Biophys Res Commun 331:761-777
- Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, et al. (2007) An attempt to prevent senescence; A mitochondrial approach Biochemistry (Moscow) 72: 1385-1396.
- Obukhova MR, Pasyukova LA, Pisarenko EG, Roginsky OI, Ruuge VA, et al. (2009) An attempt to prevent senescence a mitochondrial approach. Biochim Biophys Acta 1787: 437-461.
- Folkers K, Langsjoen P, Willis R, Richardson P, Xia LJ, et al. (1990) Lovastatin decreases coenzyme Q levels in humans. Proc Natl Acad SciUSA 87:8931-8934.
- Lehninger AL (1971) The molecular organization of mitochondrial membranes. Adv Cytopharmacol 1:199-208.
- Beal MF (2003) Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann Neurol53:S39-S47
- Kalayci M,Unal MM,Gul S,Acikgoz S,Kandemir N,et al. (2011) ofcoenzyme Q10onischemiaand neuronal damage in an experimental traumaticbrain-injury model in rats. BMC Neurosci. 29: 12-75
- Mellors A, Tappel AL (1966) the inhibition of mitochondrial peroxidation by ubiquinone and ubiquinol. J Biol Chem 241:4353-4356.
- Horecký J,Gvozdjáková A,Kucharská J,Obrenovich ME,Palacios HH,et al. (2010) Effects of coenzyme Q and creatine supplementation onbrainenergy metabolism in rats exposed to chroniccerebralhypoperfusion. Curr Alzheimer Res 8:868-875.
- Lemke M, Frei B, Ames BN, Faden AI(1990) Decreases in tissue levels of ubiquinol -9 and -10, ascorbate and alpha toco-pherol following spinal cord impact trauma in rats. Neurosci Lett 108:201-206.
- Piotrowski P, Ostrowski RP, Pankowska T, Smialek M (1998) The effect of coenzyme Q10 on lactate acidosis at the beginning of experimental cerebral ischemia in rats after the use of endothelin 1 (preliminary results) Neurol Neurochir Pol 32:1397-1404
- Lemke M, Frei B, Ames BN, Faden AI (1990) Decreases in tissue levels of ubiquinol 9 and 10, ascorbate and alpha-tocopherol following spinal cord impact trauma in rats. Neurosci Lett 108:201-206.
- Murphy MP, Smith RA (2000) Drug delivery to mitochondria: the key to mitochondrial medicine. Adv. Drug Deliv Rev 41: 235-250
- Smith RA, Porteous J, Gane CM, Murphy MP (2003) Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl Acad Sci USA 100: 5407-5412
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, et al. (2001) Selective targeting of a redox active ubiquinone to mitochondria within cells J Biol Chem 276:4588-4596
- Neuzil J (2007) Mitochondria transmit apoptosis signaling in cardiomycetes like cells and isolated hearts exposed to experimental ischemia reperfusion injury Redox Rep 12:148-162.
- Ross MF (2008) Rapid and extensive uptake and activation of hydrophobic triphenyle phosphonium Cations within cells Biochem 411:633-645
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, et al. (2005) Targeting an antioxidant to mitochondria decreases cardiacischemia reperfusion injury. FASEB J 19: 1088-1095.
- Esplugues JV, Rocha M, Nunez C, BoscaI, IbizaS,et al. (2006) Complex1 dysfunction and tolerance to nitroglycerine and approach based on mitochondria targeted antioxidant. Circ Res 99:1067-1075.
- Wainwright M, Crossley KB (2002) Methylene Blue a therapeutic dye for all seasons J Chemother14: 431-443.
- Tretter L, Horvath G, Holgyesi A, Essek F, AdamVizi V (2014) Enhanced hydrogen peroxide generation accompanies the beneficial bioenergetic effects of methylene blue in isolated brain mitochondria. Free Radic Biol Med77: 317-330.
- Oz M, Lorke DE, Hasan M, Petroianu GA (2011) Cellular and molecular actions of Methylene Blue in the nervous system. Med Res Rev 31: 93-117.
- Shen Q, Du F, Huang S, Rodriguez P, Watts LT, et al. (2013) Neuroprotective Efficacy of Methylene Blue in Ischemic Stroke: An MRI Study. PLoS ONE 8: e79833.
- Di Y, He YL, Zhao T, Huang S, Wu KW, et al. (2015) Methylene Blue Reduces Acute Cerebral Ischemic Injury via the Induction of Mitophagy. Mol Med
- Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F (2004) Methylene blue improve brain oxidative metabolism and memory retention in rats. Pharmacol Biochem. 77:175-181
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria central role of complex III. J Biol Chem 278: 36027-36031.
- Sen S, Phillis JW (1993) alpha-Phenyl-tert-butyl-nitrone (PBN) attenuates hydroxyl radical production during ischemia-reperfusion injury of rat brain: an EPR study. Free Radical Research Communications 19: 255-265.
- Kuroda S , Tsuchidate R , Smith ML , Maples KR , Siesjo BK (1999) Neuroprotective effects of a novel nitrone, NXY-059, after transient focal cerebral ischemia in the rat . J. Cereb. Blood Flow Metab19: 778-787.
- Cao X, Phillis WJ (1994) Alpha-Phenyl-tert-butyl-nitrone reduces cortical infarct and edema in rats subjected to focal ischemia. Brain Research 644: 267-272.
- Li PA, He QP, Nakamura, Csiszar KL (2001) Free radical spin trap alpha-phenyl-N-tert-butyl-nitron inhibits caspase-3 activation and reduces brain damage following a severe forebrain ischemic injury. Free Radical Biology and Medicine 31:1191-1197.
- Kotake Y, Sang H, Miyajima T, Wallis GL (1998) Inhibition of NF-kappaB, iNOS mRNA, COX2 mRNA, and COX catalytic activity by phenyl-Nbutylnitrone (PBN).Biochim Biophys Acta1448: 77-84.
- Alberts MJ (1999) Diagnosis and treatment of ischemic stroke Am J bed106: 211-221.

