Piradee Suwanpakdee1*, Napakjira Likasitthananon1, Charcrin Nabangchang1, Yutthana Pansuwan2, Siriporn Pattharathitikul3 and Boonchai Boonyawat4
1Division of Neurology, Department of Pediatrics, Phramongkutklao Hospital and Phramongkutklao College of Medicine, Bangkok, Thailand
2Phramongkutklao Hospital and Phramongkutklao College of Medicine, Bangkok, Thailand
3Division of Pediatrics, Prapokklao Hospital, Bangkok, Thailand
4Division of Genetics, Department of Pediatrics, Phramongkutklao Hospital and Phramongkutklao College of Medicine, Bangkok, Thailand
Corresponding Author:
Dr. Piradee Suwanpakdee, MD
Division of Neurology, Department of Pediatrics, Phramongkutklao Hospital and Phramongkutklao College of Medicine, 315 Ratchawithi Rd, Thung Phaya Thai, Ratchathewi district, Bangkok 10400, Thailand
Tel: +66814383634
E-mail: piradee@pedpmk.org
Rec Date: October 25, 2018; Acc Date: November 10, 2018, 2018; Pub Date: November 14, 2018
Citation: Suwanpakdee P, Likasitthananon N, Nabangchang C, Pansuwan Y, Pattharathitikul S, et al. (2018) Molecular Analysis of PANK2 Gene in Two Thai Classic Pantothenate Kinase-Associated Neurodegeneration (PKAN) Patients. J Neurol Neurosci Vol.9 No.6:275. doi:10.21767/2171-6625.1000275.
Keywords
Molecular analysis; PANK2 gene; Pantothenate kinase-associated neurodegeneration (PKAN); Thailand
Introduction
Pantothenate kinase-associated neurodegeneration (PKAN, OMIM 234220), previously named Hallervorden-Spatz syndrome, is a rare autosomal recessive neurodegeneration with brain iron accumulation (NBIA) disorder; it has an estimated prevalence of 1 to 3 per million and accounts for almost half of NBIA cases [1,2]. PKAN is characterized by progressive extrapyramidal dysfunction and iron accumulation in the brain. Brain magnetic resonance imaging (MRI) usually shows typical “eye-of-the-tiger” pattern in the globus pallidus which is caused by iron accumulation in the peripheral region (seen as hypointensity) and necrosis in the anteromedial region (seen as hyperintensity).
PKAN is classified into two main clinical phenotypes: classic and atypical, based on the age of onset, rate of progression and severity of neurological symptoms. Classic PKAN usually presents within the first decade of life (early onset) and has a rapid progression. In case of atypical PKAN, the onset is late, and the progression is slower and more variable.
PKAN is caused by a mutation in the PANK2 gene (OMIM 606157) which is located at 20p13 and spans approximately 34 kb. This gene contains 7 exons and encodes a 571-amino acid pentothenase kinase-2 (PANK2) mitochondrial enzyme [2]. PANK2 is the principal regulatory enzyme in coenzyme A (CoA) biosynthesis and is localized in the mitochondria [3]. Due to the participation of CoA in several metabolic pathways such as the citric acid cycle, sterol and steroid biosynthesis, heme biosynthesis, and amino acid and fatty acid metabolism, deficiency of CoA due to PANK2 mutations leads to diverse metabolic defects and possible mitochondrial dysfunction.
To date, more than 150 pathogenic mutations have been reported in various ethnic groups [4]. Almost all mutations were small rearrangements including missense, nonsense, splice-site mutations, and small deletions and insertions. Most of these mutations were missense mutations.
In this study, we report the clinical features, neuroimaging and molecular analysis of the PANK2 gene in two Thai patients suffering from classic PKAN.
Case Reports
Patient 1
A 12-year-old Thai boy presented with progressive dystonia since early childhood. He started walking at the age of 1 year and 8 months; at that time his mother noticed that he walked on the tips of his toes and fell frequently. When he was 5 years old, he developed dystonia which progressed exponentially. At the age of 11 years, he had dysphagia and dysarthria which were caused by mouth and tongue dystonia, respectively. He also had difficulty speaking. The symptoms of generalized spasticity became more progressive and limited his daily activities; thus, he had to quit school. Family history revealed that he was the first child of a consanguineous couple and had two healthy younger siblings: a brother and a sister.
Neurological examination revealed dystonic face, generalized dystonia, dysarthria and hyperreflexia of all limbs. T2-weighted brain magnetic resonance imaging (MRI) demonstrated symmetrical hyperintensity at the medial aspect of the globus pallidus, resulting in an “eye-of-the-tiger” appearance (Figure 1A). Owing to clinical features and the typical “eye-of-the-tiger signal” on the brain MRI, PKAN was suspected. Despite the use of benzodiazepine, baclofen, levodopa, deferiprone, and pantothenic acid, his dystonia and spasticity were not improved. He became wheelchair-bound 8 years after the onset of symptoms.
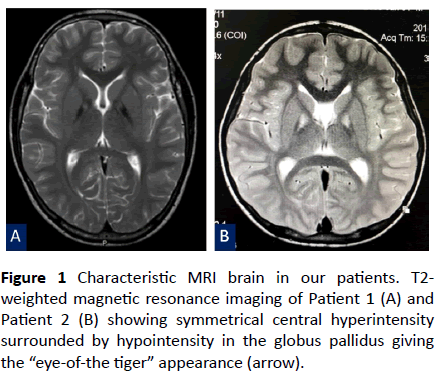
Figure 1 Characteristic MRI brain in our patients. T2- weighted magnetic resonance imaging of Patient 1 (A) and Patient 2 (B) showing symmetrical central hyperintensity surrounded by hypointensity in the globus pallidus giving the “eye-of-the tiger” appearance (arrow).
Patient 2
A 7-year-old Thai boy presented with developmental regression since he was 3 years of age. He was born to nonconsanguineous parents and was healthy until the age of 3 years. He had issues walking and fell down frequently. Furthermore, the parents had noticed a slight psychomotor developmental delay, which affected his school performance. He also had a speech disorder and could not speak long sentences. Neurological examination showed impaired memory and calculation ability, dystonic posturing, and hyperreflexia of both arms and legs. Neither weakness nor abnormal cerebellar signs were observed. Developmental regression with marked dystonia was the primary concern in this patient. Brain MRI was performed and revealed the typical “eye-of-the-tiger” signal in the bilateral globus pallidus (Figure 1B). Thus, PKAN was strongly suspected. Baclofen and deferiprone were administered but the patient’s symptoms kept progressing.
Methods
Mutation analysis of the PANK2 gene
After informed consent was obtained from the patients and their family members, genomic DNA was extracted from peripheral blood lymphocytes using commercially available kits according to the manufacturer’s protocol. All coding exons and exon-intron boundaries of the PANK2 gene were amplified through PCR using 7 pairs of primers as previously described [5]. All PCR products were purified and sequenced in both directions. Novel PANK2 mutations were investigated in 100 healthy control alleles and were analyzed through the PolyPhen-2 (https://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.jcvi.org/), and Mutation Taster (https:// www.mutationtaster.org/) softwares to predict the effect of non-synonymous variations. The reference sequences were NM_153638.3 and NP_705902.2 for PANK2 gene and PANK2 protein, respectively.
Results
A homozygous mutation of c.1475C>T (p.Ala492Val) was identified in patient 1 (Figure 2A). This mutation was located on exon 5. Segregation analysis of available family members including one younger brother, one younger sister and both parents was performed. All of them were heterozygous for the same mutation (Figure 2B). This missense mutation had been previously reported in The Human Gene Mutation Database (HGMD) (https://www.hgmd.cf.ac.uk).
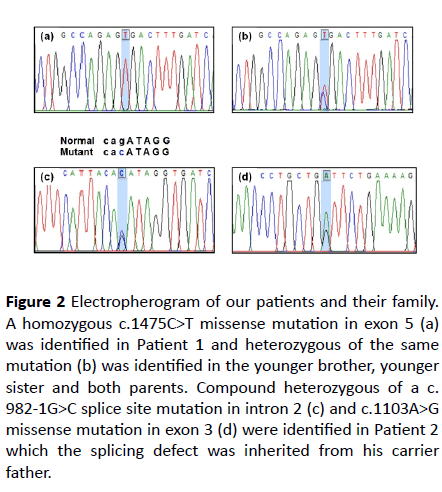
Figure 2 Electropherogram of our patients and their family. A homozygous c.1475C>T missense mutation in exon 5 (a) was identified in Patient 1 and heterozygous of the same mutation (b) was identified in the younger brother, younger sister and both parents. Compound heterozygous of a c. 982-1G>C splice site mutation in intron 2 (c) and c.1103A>G missense mutation in exon 3 (d) were identified in Patient 2
which the splicing defect was inherited from his carrier father.
Compound heterozygous genotypes of a known c.982-1G>C (IVS2-1G>C) splice site mutation in intron 2 and a novel c. 1103A>G (p.Asp368Gly) missense mutation in exon 3 were identified in patient 2 (Figures 2C and 2D). The former splicing defect was inherited from the father. Unfortunately, genomic DNA from the mother was not available. Concerning this novel missense mutation, Polyphen-2 and Mutation Taster software predicted the pathogenicity to be “possibly damaging” and “disease causing”, respectively. The SIFT software predicted the pathogenicity to be “tolerated.” However, this missense mutation was not identified in 100 healthy control alleles, the HGMD, the 1000 Genome Project database (https:// www.internationalgenome.org/), and the Exome Aggregation Consortium (https://exac.broadinstitute.org/).
Discussion
PKAN is caused by mutations in the PANK2 gene which encodes a pantothenate kinase-2 (PANK2) mitochondrial enzyme. PANK2 is the first and rate-limiting enzyme in coenzyme A (CoA) biosynthesis. Deficiency of PANK2 results in both CoA deficiency, which affects cellular energy production and accumulation of cysteine-containing molecules, which in the presence of iron may cause rapid auto-oxidation leading to free radical generation and extensive cellular damage [1,2]. Since PANK2 is expressed ubiquitously in basal ganglia and retina, iron accumulation in the basal ganglia especially in the globus pallidus and retinopathy are cardinal features for this disorder [2]. The two main forms of clinical presentation are: classical, which start in early childhood with rapid progression of symptoms including dystonia, dysarthria, rigidity, and cognitive decline; and atypical, which has later onset (after the age of 10 years) and slower progression. Unlike in the classic presentation, in the atypical presentation, motor disorders are fewer, but there are more psychiatric symptoms and speech disorders.
We reported two classic PKAN Thai patients presented with progressive dystonia. Brain MRI revealed the typical “eye-ofthe- tiger” signal and PANK2 mutations were identified in both patients. Typically, the main clinical feature of classic PKAN is progressive dystonia, but the type of dystonia differs among Asians and Caucasians. Previous studies mention that cranial dystonia is much less common in Asians than in Caucasians in both classic and atypical PKAN patients [6]. In contrast, atypical PKAN patients in Asia tend to have segmental dystonia more than Caucasians. Moreover, they also pointed out that Caucasians with PKAN have more complex presentations than Asians. High prevalence of pyramidal signs, mental impairment and parkinsonism were observed in Caucasian patients, whereas dysarthria was frequently noticed in classic PKAN patients of Asian origin. Our study is in line with these studies, the main clinical features of classic Thai PKAN are similar to those in Asian PKAN. The patients presented with progressive generalized dystonia and dysarthria and no sign of parkinsonism is observed.
Ethnicity also plays an important role in genetic analysis. To date, more than 150 pathogenic mutations including missense, nonsense, splice-site mutations, and small deletions and insertions have been identified throughout the PANK2 gene and have been reported in PKAN patients from different populations [4,6-10]. Two missense mutations including c. 1475C>T (p.Ala492Val) and c.1103A>G (p.Asp368Gly) and one splice site mutation, c.982-1G>C (IVS2-1G>C), were identified in our study. The c.1475C>T missense mutation has been previously reported in HGMD with no clinical information on the patient. In this study, homozygosity of this mutation resulted in classic PKAN as demonstrated in our first patient. The c.1103A>G is a novel missense mutation and has never been reported in previous studies [4,6-10]. Polyphen-2 and Mutation taster softwares predicted the pathogenicity of this mutation to be possibly damaging and disease-causing, respectively, whereas, SIFT predicted the pathogenicity to be tolerated. However, this mutation was neither identified in 100 healthy control alleles, nor in other large genomic databases. Although the maternal genomic DNA was not available, the paternal and the patient’s DNA suggested the possibility of autosomal recessive inheritance pattern. Unfortunately, further vlidation cannot be performed at our institution. The c. 982-1G>C intronic mutation has been previously reported in the first and only report of a Thai classic PKAN patient who was homozygous for this intronic mutation [11]. This finding suggests the ethnic specificity of this recurrent mutation in Thai populations.
Although the PANK2 mutation has been identified in all patients with classic PKAN, as demonstrated in our study, the mutation can be identified in only one-third of atypical PKAN patients [10]. Nevertheless, when PKAN is suspected, genetic testing is recommended to confirm the diagnosis in affected patients and presymptomatic diagnosis in other family members. In addition to diagnostic confirmation of suspected disease, molecular analysis is also important for carrier testing, prognosis, and facilitation of prenatal or pre-implantation genetic diagnosis.
Conclusion
Our study demonstrates the clinical presentations and molecular analysis of two Thai patients with classic PKAN. Generalized dystonia and dysarthria were the main clinical features that differed from classic PKAN in Caucasians. In addition, two unique missense mutations and one recurrent splicing defect were also identified in Thai patients with classic PKAN.
Acknowledgments
None.
Funding
None.
Competing and Conflicting Interests
There are no conflicts of interest.
23735
References
- Akcakaya NH, Iseri SU, Bilir B, Battaloglu E, Tekturk P, et al. (2017) Clinical and genetic features of PKAN patients in a tertiary centre in Turkey. Clin Neurol Neurosurg 154: 34-42.
- Ghafouri-Fard S, Yassaee VR, Rezayi A, Hashemi-Gorji F, Alipour N, et al. (2016) A Novel Nonsense Mutation in PANK2 Gene in Two Patients with Pantothenate Kinase-Associated Neurodegeneration. Int J Mol Cell Med 5: 255-259.
- Hartig MB, Hörtnagel K, Garavaglia B, Zorzi G, Kmiec T, et al. (2006) Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol 59: 248-256.
- Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, et al. (2003) Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 348:33-40.
- Hortnagel K, Prokisch H, Meitinger T (2003) An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum Mol Genet 12:321-327.
- Lee CH, Lu CS, Chuang WL, Yeh TH, Jung SM, et al. (2013) Phenotypes and genotypes of patients with pantothenate kinase-associated neurodegeneration in Asian and Caucasian populations: 2 cases and literature review. Scientific World Journal 2013: 860539.
- Pellecchia MT, Valente EM, Cif L, Salvi S, Albanese A, et al. (2005) The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology 64: 1810-1812.
- Perez-Gonzalez EA, Chacón-Camacho OF, Arteaga-Vázquez J, Zenteno JC, Mutchinick O (2013) A novel gene mutation in PANK2 in a patient with an atypical form of pantothenate kinase-associated neurodegeneration. Eur J Med Genet 56: 606-608.
- Schneider SA, Hardy J, Bhatia KP (2012) Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord 27: 42-53.
- Trachoo O, Satirapod C, Panthan B, Sukprasert M, Charoenyingwattana A, et al. (2017) First successful trial of preimplantation genetic diagnosis for pantothenate kinase-associated neurodegeneration. J Assist Reprod Genet 34: 109-116.
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, et al. (2001) A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 28: 345-349.

