Keywords
|
| Signaling cascades; Biological networks; Drug discovery; Molecular phenotyping |
Introduction
|
| Before two independent draft versions of the human genome were published in 2001, prominent scientists predicted understanding of the genetic basis of the majority of human disease and rational drug development once the human genome is deciphered [1,2]. More than a decade later hundreds of human genome sequences have been solved and deposited in public domain databases. Since, genome wide association studies (GWAS’) have been conducted in controlled human populations to associate genotypes with target genes or loci causing disease. Today, the results related to novel therapies are rather disappointing not fulfilling the expectations. On the other hand, novel and emerging technologies for genome analysis have changed the way we approach the underlying molecular mechanisms for human disorders. As result, comprehensive gene expression databases with signatures from many human tissues became available. In parallel, complementary efforts in the field of proteomics led to recently published databases containing quantitative and qualitative expression data for all known proteins in many human tissues [3,4]. In 2004, a computational analysis of the human genome allowed mapping of enzymatic activities to predicted metabolic pathways [5] thereby complementing the famous biochemical pathway poster published more than 20 years ago (https:// www.roche.com). |
| At the same time robust and affordable microarray platforms were developed allowing generation of complex tissue gene expression databases from humans and relevant animal models. Among other things, these approaches led to discovery of gene expression signatures comprising relatively small sets of genes predictive for disease types or pharmacological responses. In 2006, the LuminexFlexMAP (LMF) method was published that allowed customized discovery of disease relevant tissue gene expression signatures in large numbers of samples coming for example from a clinical phase III study [6]. The availability of reliable high-throughput tools in virtually all omics-related scientific disciplines resulted in a variety of databases mostly derived from publications where data release into the public domain became mandatory. |
| This wealth of shared information led to the development of numerous interactive pathway databases with user friendly interfaces. The reference version of KEGG PATHWAY for instance is defined as “database of biological systems that integrates genomic, chemical and systemic functional information” and a number of extensions such as KEGG DRUG or KEGG DISEASE have been added since [7]. PID, the pathway interaction database has been created in collaboration with the Nature Publishing Group and adds literature data for interactions to the network level centric representation of KEGG or REACTOME [8]. REACTOME contains the participation data of 7088 human proteins in 6744 reactions published in more than 15,000 articles with PubMed links. The ENCODE consortium aims at identification of all functional elements in the human genome and the output is powered by Nature’s Encode Explorer for data and literature mining (https://www.genome. gov/encode/). SAMNetWeb is a recent tool that provides a user friendly interface for data integration from multiple sources and experiments [9]. |
| Recently, pathway and network centric approaches for drug discovery and safety were published [10,11]. Such strategies were applied for identification of pathways and mechanisms related to cancer treatment, drug resistance or disease progression based on functional phenotypes of 100 cancer cell lines treated with RNA interference shRNA libraries [12]. This screen yielded unique or common network motifs defined as coherent groups of functionally related genetic regulators. In addition, this approach enabled discrimination between on- and offtarget effects of shRNA mediated interference which is common for this screening strategy and complicates data interpretation. |
| Moreover, DNA sequencing based high-throughput screening (HTS) identified the mode of action and signaling cascades of anticancer drugs [11]. Interestingly, a group of cardiac glycosides activates of the androgen receptor signaling pathway required for prostate cancer therapy. For example, compounds like Peruvoside efficiently inhibit cell proliferation in vitro thereby opening the door for novel cancer treatment modalities. This example shows that novel medicines can be efficiently discovered based on pathway modulation without prior knowledge of the direct drug target. |
| To uncover adverse side-effects of new drug candidates associated with transcription, toxicogenomics was introduced in the field of drug safety and toxicity testing about 15 years ago. Traditionally, data analysis was performed at the gene level for individual compounds and these small-scale experiments did not deliver the expected toxicity signatures. However, an integrated network based analysis of the public TG-GATEs database [13] containing pathological records, transcriptional profiles and cell based readouts for 170 compounds resulted in identification of four genes (EGR1, GDF15, FGF21 and ATF3) that are associated with toxicity regardless of the compound class or molecular target. The products of these genes are genetic regulators that control signaling cascades of stress response, apoptosis, lipid metabolism and immune responses [14]. Interestingly transcriptional modulation of these genes occurs two hours after exposure to the toxic insult signifying favorable features of predictive toxicity biomarkers. |
| This study led to the concept of molecular phenotyping using pathway reporter genes such as EGR1, GDF12, FGF21 and ATF3 for the toxic “molecular phenotype”. This important finding triggered a global search for pathway reporter genes covering 154 human signaling networks regulating major cellular processes of human cells and organs (hormone and neuropeptide signaling, stress response, fatty acid metabolism, nucleic acid metabolism, energy and drug metabolism, DNA damage and apoptosis, immune and inflammation response, growth and transformation signaling pathways, cell differentiation). This search of literature, private and public databases resulted in a final set of 917 candidate pathway reporter genes or regulators with the desired properties such as transcription- or cell growth factors [15]. Following in silico validation of the panel, we applied a digital deep-sequencing based RNA quantification technology termed RNAAmpliSeq for biological validation of the panel pathway reporter genes [15]. We have chosen several time points during differentiation of human stem cells into young cardiomyoctes because we anticipated modulation of most pathways during development of a polypotent precursor cell into a highly specialized. Indeed we detected modulation of 151 pathways during this differentiation process at specific time points. The three unmodulated pathways regulate processes of the central nervous system. Application of sequencing based RNA quantification methods such as AmpliSeq or HTS [11] for molecular phenotyping has the advantage that these methods cover a large dynamic range, single-molecule sensitivity and no background due to sequence based transcript quantification. |
| A contemporary drawback of deep-sequencing based approaches is lack of efficient automation by main technology providers and high cost. For high-throughput screening projects fluorescence based multiparallel technologies such as the L1000 hybridization method are better suited for screening several thousand of samples at the cost of lower sensitivity and precision. Analogous to pathway reporter genes described above, the LINCS expression libraries were constructed using responses of about 1000 landmark-genes selected from different signaling cascades [16]. This approach was successfully used for the generation gene expression signature libraries defining distinct biological processes and responses (https://www.maayanlab.net/LINCS/ LCB). |
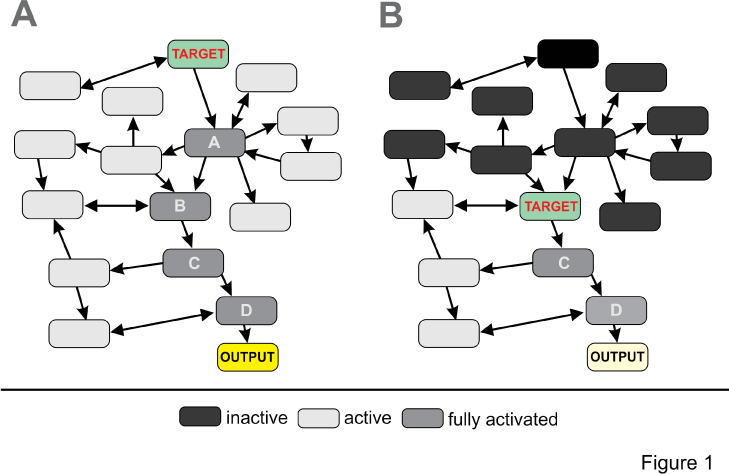
| Regardless of the technology chosen, these recently published pathway centric-approaches allow for molecular phenotype based drug screens without information of the actual drug target. Figure 1 shows two virtual outcome scenarios of pathway (panel A) versus target (panel B) based screening approaches. In the pathway centric approach, the key regulator of the entire pathway is modulated by an agonist or antagonist resulting in activation of the entire network resulting in potent physiological output such as inhibition of cell proliferation typical for anti-cancer drugs. In the target based approach, a target downstream of the key regulator is modulated and as a result partial activation of the same network occurs resulting in less potent output. |
| Molecular phenotype based screens open the possibility to repurpose approved drugs for other therapeutic indications as long as they modulate the same network defined ideally by an established “gold standard” drugs such as the statins or metformin for type II diabetes. This new, pathway based strategy for extending therapeutic applications of marketed drugs would significantly reduce costs and time associated with professional drug development. Furthermore, drugs with promiscuous target specificity also referred to as off-target activity, might be used for novel therapeutic indication provided they co-modulated the networks defined by gold-standard drugs. |
| A recently published chemical proteomic study provides a nice example for this paradigm based on kinase inhibitors frequently used in cancer therapy [17]. All kinase inhibitors block entry of ATP into the binding pocket of kinases which implies that target selectivity is an issue for this class of compounds. Using an elegant combination of affinity binding to beads, competition with kinase inhibitors and mass spectrometry analysis, the target selectivity of 9 marketed kinase inhibitor drugs and their affinities (Kd) was analyzed in a lysate of seven tumor cell lines expressing the entire human kinome. As it turned out, none of the compounds was monospecific and interestingly offtarget binding of Dasatinib for instance to several other tyrosine kinase occurred at similar affinity as binding to the targets kinases BCR-ABL and SRC. Consequently, Dasantinib can be repurposed for therapies were activation of one of the off-target kinases correlates with the disease phenotype. This approach can be further refined by network based molecular phenotyping and identification of the downstream signaling cascades concordant with model shown in Figure 1. |
| Finally, molecular phenotyping using pathway reporter genes results in a significant reduction of data volumes of a given experiment and allows for use of standard software packages originally developed for microarray profiling for example. More important is the circumstance that differential gene expression data at the pathway level enable mechanistic data analysis and reliable design of functional studies confirming the hypothesis as recently shown for a liver toxin [10]. |
Acknowledgements
|
| I wish to thank Drs. Jitao David Zhang and Martin Ebeling for their contributions and editing of this review. |
Figures at a glance
|
 |
| Figure 1 |
|


