Zylfije Hundozi1, Granit Xhiha1*, Jehona Rrustemi1, Bujar Gjikolli3 and Fisnik Jashari3
1Department of Neurology, University Clinical Center of Kosovo, Prishtina, Kosovo, 10000, Albania
2Department of Radiology, University Clinical Center of Kosovo, Prishtina, Kosovo, 10000, Albania
- *Corresponding Author:
- Granit Xhiha
Department of Neurology
University Clinical Center of Kosovo, Prishtina, Kosovo, 10000
Albania
Tel: +37744385682;
E-mail: xgranit@hotmail.com
Received date: April 13, 2017; Accepted date: May 19, 2017; Published date: May 22, 2017
Citation: Hundozi Z, Xhiha G, Rrustemi J, Gjikolli B, Jashari F (2017) Neuromyelitis Optica with a Good Response After Low Doses of Corticosteroid Therapy: A Case Report. Int J Drug Dev & Res 9: 08-09
Keywords
Neuro myelitis optica; Transverse myelitis; Aquaporin-4 antibodies; Corticosteroid therapy
Introduction
Neuromyelitis optica, also known as Devices disease or Devices syndrome is an immune mediated chronic inflammatory disease of central nervous system. Clinically, it presents with optic neuritis (ON) and myelitis, often characterized by poor or no recovery [1]. It can be monophasic or recurrent. The prevalence and incidence of Devices disease has not been established, because it is underdiagnosed and also confused with sclerosis multiplex. Its prevalence is estimated to range from less than 1 to 4.4/100.000 in western World [2]. More women than men have NMO (ratio 9:1, compared to Sclerosis multiplex which is 2:1) [3]. Specific etiological causes of NO have not been identified, although connective tissue disorders, tuberculosis, and acute disseminated encephalomyelitis have been associated with it. Although originally considered as variation of multiple sclerosis, clinical radiological pathological and especially immunological data have led to a novel definition of this clinical entity [4]. It is currently considered autoimmune disease (autoimmune astrocytopathy or autoimmune astrociticchannelopathy) based on presence of autoantibodies against AQP4 [5]. NMO-IgG seropositive patients with a history of optic neuritis or transverse myelitis who do not fulfill all of the clinical criteria are classified as having NMO spectrum disorder but are clinically treated identically as definite NMO. The clinical hallmarks of NMO are acute neuritis that is often bilateral and transverse myelitis that is often longitudinally extensive. Commonly reported symptoms include unilateral and bilateral loss of visual acuity, ocular pain, and severe paraplegia, a symmetric sensory level, bladder dysfunction, and paroxysmal tonic spasm of the trunk and limbs, and Lermitte´s phenomenon [6].
Magnetic resonance imaging (MRI) reveals involvement of more than three vertebral segments, which is synonymous with longitudinally extensive transverse myelitis (LETM) [7-9].
Case Presentation
A 54 years old women was hospitalized in the department of Neurology with loss of vision and pain on the right eye, progressive weakness and numbness on her right hand, headache, vertigo, vomiting, and bladder dysfunction. She had anxiety accompanied by poor endurance. Complaints started two days before admission in Neurologic department. This was second hospitalization in department of Neurology. First time she had left side hemiparesis. Family history was unremarkable. During Neurological assessment muscle power grade 3/4 on the Medical Research Council (MRC) scale on right hand with painful spasticity and brisk deep tendon reflexes. Sensory deficit to fine touch and pressure on the right hand. An afferent pupillary defect on right eye, and eye pain worsened with eye movement. She had urinary incontinence and urinary catheter was placed in situ.
The full blood count showed the following: WBC: 10.2-10³/ mm³, RBC: 4.92-10³/mm³, lymphocites: 24, 1%, Monocites: 5, 9%, Granulocites: 70%, HGB: 13.1 g/dl, HTC: 41.0%, PLT: 356-10³/mm³, PCT: .230%. It was detected a high level of autoantibodies directed to AQP4 qualitative ++ and quantitative (RIA): 2, 4 nM (normal values ranged 0.0 to 0.4 nM). A lumbar puncture was performed and showing normal glucose and protein level lactate level, with WBC count of 5 cells/uL, IgG, IgA and IgM CSF/serum ratios were normal. The presence of oligoclonal bands in liquor was negative. Brain MRI revealed hypertrophy of right optic nerve (Figure 1) and cervical spine MRI reviled multiple hyper intense signals involving cervical cord levels C1, C2, C3, C4 and C6 on T2–weighted images (Figure 2).
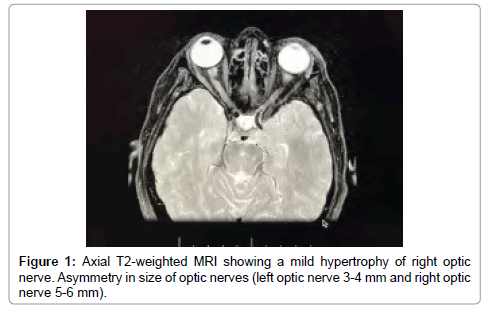
Figure 1: Axial T2-weighted MRI showing a mild hypertrophy of right optic nerve. Asymmetry in size of optic nerves (left optic nerve 3-4 mm and right optic nerve 5-6 mm).
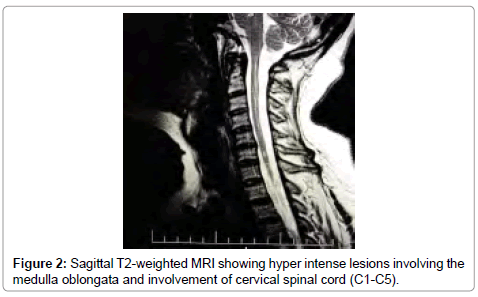
Figure 2: Sagittal T2-weighted MRI showing hyper intense lesions involving the medulla oblongata and involvement of cervical spinal cord (C1-C5).
Since in the first few days after admission the diagnosis of NMO was not yet confirmed, the treatment was started with low-dose intravenous methylprednisolone of only 120 mg daily for 5 days and was followed by an oral prednisone taper 60 mg every other day together with azathioprine 2.5 mg/kg/day. She had a good response with her vision improved after second day of therapy administration.
Discussion
Neuromyelitis optica (NMO) is severe idiopathic autoimmune disorder of the central nervous system. For reasons not completely understood, NMO preferentially affects the optic nerves and spinal cord causing relapses of optic neuritis and transverse myelitis associated with a long lesion in the spinal cord, typically extending three or more spinal segments (longitudinally extensive transverse myelitis. NMO was considered as variant of multiple sclerosis, but now is regarded as a distinct disease entity since Aquaporin-4 antibodies were first described in 2004.
Our case presentation shows a detailed medical history, physical examination, laboratory investigations; brain MRI, cervical and thoracic spine MRI together with serologic evaluation. All findings fulfill criteria for diagnosis of NMO: optic neuritis, acute myelitis, NMO-IgG seropositive status, contiguous spinal cord MRI lesions extending over ≥ 3 segments in the context of an acute myelitis attack. Our focus was on not only on primary symptoms but also on symptoms that could suggest alternative diagnosis or concomitant autoimmune disorders which are frequently present in patients with AQP-4-Ab positive NMO but finally they were excluded. There are many case reports in literature of NMO associated with another autoimmune condition such as autoimmune thyroiditis, myasthenia gravis and Sjorgen syndromes [10-12].
Following an attack of NMO, high-dose IV Methylprednisolone (1 g for 5 days) is considered standard treatment and should be initiated as soon as it is clear that a patient is having an attack [8,13]. Long-term immunosuppressive treatment should be initiated once the diagnosis of NMO has been confirmed to prevent relapsing course in most cases [11]. Azathioprine and Rituximab are currently the most widely used first line therapies in NMO [1].
In our case reported here, low-dose of corticosteroids was started on the second day after admission. She was treated with Methylprednisolone 120 mg IV for 5 days and was followed by an oral prednisone taper every other day. For long term relapse prevention, we have started Azathioprine 2.5 mg/kg/day. She had a good response after second day of therapy administration, with her vision improved and also pain in the eye diminished. To our knowledge, it is only one case of NMO reported in the literature, which showed improvement after low corticosteroid doses [14-16].
Conclusion
NMO is diagnostic challenge in every-day clinical neurologic practice because of similarity with multiple sclerosis. Early differentiation between NMO and MS is particularly important because of course of NMO is more severe and treatment strategies for attack preventions are different. Disease modifying therapies for NMO have not been rigorously studied; we have reported a case with good response after treatment with low doses of corticosteroid therapy.
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
19511
References
- Trebst C, Jarius S, Berthele A, Paul F, Schippling S, et al. (2014) Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 261: 1-16.
- Jacob A, Panicker J, Lythgoe D, Elsone L, Mutch K, et al. (2013).The epidemiology of neuromyelitisoptica amongst adults in the Merseyside county of United Kingdom. JNeurol 260:2134-2137.
- Wingerchuk DM (2009) Neuromyelitisoptica: effect of gender.J Neurol Sci286:18-23.
- Beiruti K, Saleh AS, Daitzman M, Shahien R (2016) NeuromyelitisOptica Spectrum disorders: A Case report.JMult Scler 3: 192.
- Lucchinetti CF, Guo Y, Popescu BFG, Fujihara K, Itoyama Y, et al. (2014) The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol 24: 83-97.
- Kimbrough DJ, Fujihara K, Jacob A, Lana-Peixoto MA, Leite MI, et al. (2012) Treatment of neuromyelitis optica: review and recommendations. MultScler Relat Disord 1: 180-187.
- Roy U, Saini DS, Pan K, Pandit A, Ganguly G, et al. (2016) Neuromyelitis optica spectrum disorder with tumefactive demyelination mimicking multiple sclerosis: a rare case. Front Neurol7.
- Matiello M, Weinshenker BG (2014) Multiple Sclerosis and CNS inflammatory Disorders. John Willey &sons.
- Jarius S, Paul F, Franciotta D, de SezeJ Münch C, Salvetti M, et al. (2012) Neuromyelitis spectrum disordes in patients with myasthenia gravis: ten new aquaporin -4 antibody positive cases and review of the literature. MultScler 18: 1135-43.
- Sudulangunta Rao S, Sodalanguta Babu M, Khorram H, Gonivada J, Noroozpour Z, et al. (2015) Autoimune thyroiditis associated with neuromyelitisoptica (NMO). Ger Med Sci 13: 22.
- Carvalho DC, Tironi TS, Freitas DS, Kleinpaul R, Talim NC, et al. (2014) Sjögren syndrome and neuromyelitisoptica spectrum disorders coexist in a common autoimmune milieu. ArqNeuropsiquatr 72: 619-24.
- Wingerchuk DM, Weinshenker BG (2008) Neuromyelitisoptica. Cur Treat Options Neurol10:55-66.
- Jarius S, Aboul-Enein F, Waters P, Hauser A, Berger T, et al. (2008) Antibody to aquaporin-4 in the lon- term course of neuromyelitisoptica. Brain 131:3072-3080.
- Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, et al. (2011) Azathioprine tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 77: 659-666.
- Bichuetti DB, de Oliveira EML, Oliveira DM, de Souza NA, Gabbai AA (2010) Neuromyelitis optica treatment: analysis of 36 patients. ArchNeurol 67: 1131-1136.
- Mitsuyama T, Yoneyama T, Suzuki S, Kawamata T (2012) Neuromyelitisoptica spectrum disorder: a case report. No ShinkeiGeka 40:337-42.


