Key words
Diabetes mellitus; cognition; memory; neuron specific enolase
Abbreviations
BDI-II, Beck Depression Inventory 2nd edition; BMI, body mass index; CNS, central nervous system; DM, diabetes mellitus; DBP, diastolic blood pressure; DSM–IV Diagnostic and Statistical Manual of Mental Health Disorders, 4th edition; ERPs, event related potentials; FBG, fasting blood glucose; HbAIc, glycolysated hemoglobin; HDL-c, high density lipoprotein cholesterol; HOMA-IR, homeostatic model assessment to quantify insulin resistance; HTN, Hypertension; IR, insulin resistance; LDL-c, low density lipoprotein cholesterol; MMSE, Mini-mental State examination; NSE, neuron specific enolase; PBG, post-prandial blood glucose; T2DM, type 2 diabetes mellitus; TC, total cholesterol; TG, triglycerides; SBIS, Stanford Binet Intelligence Scale; SBP, systolic blood pressure; WMS-R, Wechsler Memory Scale- Revised
Introduction
Type 2 diabetes mellitus (T2DM), formerly known as noninsulin- dependent diabetes mellitus (NIDDM) or adult onset diabetes, comprises 90% of diabetes mellitus (DM). T2DM is characterized by abnormalities in carbohydrate and fat metabolism, chronic hyperglycemia, insulin resistance and a relative insulin secretion defect (1). T2DM is often associated with long-term consequences or serious adverse effects affecting many tissues and organs such as the heart, retina, kidney and nervous system (central and peripheral nervous systems) (2). These long-term consequences of chronic hyperglycemia are an important health care issue in view of the growing obesity, diabetes, sedentary life and metabolic syndrome (which is characterized by visceral obesity, elevated fasting blood sugar, elevated triglycerides, a decrease in high-density lipoprotein cholesterol levels and high blood pressure) (3).
DM is also associated with an increased risk of poor cognition, progressive memory dysfunction, dementia and neurodegeneration. Decrease in processing speed, verbal memory, psychomotor efficiency, learning, intelligence and executive functioning were observed in population-based studies of T2DM (4-8). DM-induced cognitive decline is likely of multifactorial etiology through multiple mechanisms which include: abnormalities of blood glucose (9) blood lipid (10,11), blood pressure (12-14), insulin resistance (IR) (3,15), hypoglycemia (16), chronic complication as micro- and macro- vascular complications (or diabetic vasculopathy) (17), dysregulation of limbic-hypothalamic-adrenal pituitary axis (LHPA) (stress response) (18-21), advanced glycation end products, inflammatory cytokines, oxidative stress (22,23) and diabetes-related depression (24-26). However, the possibility of the direct toxic effect of chronic hyperglcycemia has to be considered as a cause of T2DM-related cognitive impairment. The increase in neuronal vulnerability to death and apoptosis may have additive (which means an augmented net effect through action on the same pathway) or synergistic effects (which means an augmented net effect through action on two different pathways) to the vascular complications of DM.
During the last years, the possibility of evaluating brain damage/ activity through quantification of neuronal derived proteins (such as neuron specific enolase or NSE) in peripheral samples has gained appropriate attention in clinical and experimental settings. NSE is a cytoplasmatic glycolytic pathway enzyme and the γγ isoform is mainly neuronal. NSE is found in neurons and neuro-endocrine tissue and it is elevated in the blood circulation after death rate of these cells (27,28). Several studies have shown higher serum and cerebrospinal fluid (CSF) levels of NSE and also their over-expression increases the vulnerability to neurodegeneration, cerebral hypoxic-ischemic injury and traumatic brain injury (27,29). Accordingly, in T2DM, the presence of metabolic abnormalities, disturbance of vascular reactivity, hypoxia, disturbance of blood-brain barrier (BBB) permeability and excitotoxic process make the quantification of neuronal derived proteins (or NSE proteins), a sensitive and direct biomarker of brain damage as well as its related neurological and neuropsychological outcome.
Aim of work
In this study, we aimed to investigate the direct brain involvement of chronic hyperglycemia. We included a homogeneous group of T2DM patients to adjust for numerous confounders which may affect cognition. The following markers of brain involvement were investigated: 1) cognitive functions: were assessed using a battery of sensitive psychometric testing, 2) event related potentials (ERPs), a neurophysiological analogue of cognitive function, and 3) serum concentrations of NSE, a sensitive marker of neuronal damage. Correlations were done between scores of cognitive testing, ERPs variables and NSE protein levels.
Patients and methods
Patients
This cross-sectional study included 57 patients with T2DM. Patients were randomly recruited from the Internal Medicine and Neurology departments of Assiut University Hospital, Assiut, Egypt. The diagnosis of DM was made according to the World Health Organization Expert Committee on DM, Geneva: WHO (30). For the diagnosis of DM, one or more of the following criteria has to be fulfilled: 1) a fasting blood glucose (FBG) value >125 mg/dl on two separate occasions; 2) a two hours glucose (or post-prandial blood glucose or PBG) value >200 mg/dl during a 75 gram oral glucose tolerance test; or 3) a prior diagnosis of T2DM, and being treated with hypoglycemic agents and/or diet and exercise. Forty age-, sex-, socioeconomic status- and educationally- matched subjects were included in this study as healthy controls for comparison. Control subjects were recruited from the general population. The protocol of this study was in conformity with the local ethical guidelines of Assiut University hospital and informed written consent was obtained from each participant. All study participants underwent a standardized interview questionnaire regarding vascular risk factors. Excluded were subjects with: 1) known medical illness other than T2DM and its associated dyslipidemia, hypertension, IR and obesity), or primary neurological or psychiatric disease (other than diabetes- related depression); 2) history of hypoglycemic coma or diabetic complications (other than peripheral neuropathy) as nephropathy, retinopathy, etc; 3) history of transient ischemic attacks, cerebrovascular stroke or epilepsy; 4) previous serious head injury; 5) any sensory or motor disorder that would preclude psychological testing (as blindness or deafness); 6) regular treatment with any medications other than insulin and/or hypoglycemic drugs, or medication known to have psychoactive effects such as benzodiazepines, betaadrenoceptor antagonists, steroids, major tranquillizers and antidepressants; 7) drug or alcohol abuse; and 8) smokers.
Data collection
Demographic and clinical data were collected as follow: age, gender, systolic blood pressure (SBP), diastolic blood pressure (DBP), weight, height, body mass index (BMI). Weight was measured to the nearest 0.1 kg on a calibrated balance beam scale. Height was measured to the nearest 0.5 cm by a tape measure. BMI was calculated using this formula (31): weight (kg)/height (m)2. According a subject defined as normal if BMI ≥20 - ≤25 kg/m2, overweight if BMI >25 - ≤30 kg/m2, obese if BMI >30 - ≤35 kg/m2 and morbidly obese if BMI >35 kg/ m2. Hypertension (HTN) was defined according to the National Cholesterol Education Program (NCEP) guidelines (32) in presence of: 1) SBP: ≥130 mmHg, 2) DBP: ≥85 mmHg, or 3) use of anti-hypertensive medication. Dyslipidemia was also defined according to NCEP in presence of: 1) statin treatment, 2) triglycerides (TG): ≥150 mg/dl, or 3) high density lipoprotein (HDL): ≤40 mg/dl for men and ≤50 mg/dl for women. Information on smoking habit was obtained by questionnaire and patients were divided into smokers (present or former) and non-smokers (when they never smoked regularly).
Specimen collection and analysis
Venous blood samples were drawn from patients at 8.00 am. Routine hematology tests were done and included: complete blood count (CBC), renal function, lipogram [serum total cholesterol (TC), TG, low density lipoprotein cholesterol (LDL-c), high density lipoprotein cholesterol (HDL-c)] and uric acid. Serum levels of TC, TG, LDL-c and HDL-c were measured by enzymatic colourimetric method using the autoanalyzer Hitachi 911 (Boehinger, Mannheim, USA). Serum uric acid was determined by colorimetric US plus kit, supplied by Roche diagnostics, (GmbH, D-68298 Mannheim, USA). Plasma levels of FBG insulin, and glycolysated hemoglobin (HbA1c) were assessed after an overnight fast. The patients received a standard lunch (light balanced diet: 600 kcal, 35% protein, 30% fat, 35% carbohydrates). Two hours after meal, 3 ml blood samples were withdrawn from all participants for estimated of PBG level. Insulin was determined in duplicate using an enzyme-linked immunosorbent assay (ELISA) (Diagnostic Systems Laboratory, Webster, TX. USA). IR was calculated using the homeostasis model assessment (HOMA-IR) equation formula as follow: HOMA-IR = Fasting insulin (uU/mL) multiplied by fasting glucose (mmol/L) divided by 22.5. Patients were consider to have insulin resistance if HOMA-IR ≥2.6 (33). NSE level was evaluated in serum samples by UBI MAGIWEL NSE enzyme linked immunosorbant assay kits (ELISA) (UBI United Biotech inc, Cat. No.: CM-901, Mountain view, CA 94041, www.unitedbiotech.com). This is a solid phase quantitative ELISA that uses a monoclonal antibody specific for NSE (34). Reactions and quantification were performed in duplicate as described by the manufacturer. Internal software and controls provided by the manufacturer allow controlling the quality of assay.
Cognitive assessment
Cognitive functions were assessed independently for each participant by two experienced psychologists and under supervision of the psychiatrist, using a set of standardized Arabic translated neuropsychological tests which are sensitive for mild cognitive impairment and covering different cognitive domains. They included: MMSE (35,36), SBIS (4th edition) (37,38) and WMS-R (39). From SBIS, we selected vocabulary and comprehension for assessment of verbal reasoning, pattern analysis for assessment of visual reasoning, quantitation for quantitative reasoning, and bead memory and memory for sentences for short-term memory. From WMS-R, we tested digit forward, digit backward, mental control, associate learning, logical memory and visual reproduction.
Event-Related Potentials (ERPs) testing
Before examining ERPs, all participants underwent basic audiological testing (Amplaid Model 720, Milan, Italy). Testing for ERPs was done on a separate day after completion of neuropsychological testing (Neuropack S1 EMG/EP measuring system, MEB-9400 (Nihon Kohden, Japan). ERPs are series of scalp waves that are extracted from the electroencephalogram (EEG) by time domain analysis and averaging of EEG activity following multiple stimulus repetitions. They were elicited with an auditory discrimination task paradigm by presenting a series of biaural 1000 Hz (standard) versus 2000 Hz (target) tones at 70 dB with a 10 millisecond rise/fall and 40 millisecond plateau time. P300, the late component of ERPs was obtained. Latencies and amplitudes (peak to peak) of P300 component of ERPs were measured. P300 is believed to index stimulus significance and the amount of attention allocated to the eliciting stimulus event, being maximal to task-relevant or attended stimuli and being absent or small to task-irrelevant or unattended stimuli (40).
Psychological evaluation
Standardized psychiatric interview was done by applying the Diagnostic and Statistical Manual of Mental Health Disorders, 4th edition (DSM–IV) criteria for the diagnosis of depression (41). A differentiation between clinical depression and depressive symptoms was made throughout this work. The Arabic version (42) of the Beck Depression Inventory, 2nd edition (BDI-II) (43) was used for assessment of the severity of depressive symptoms. BDI–II items are in alignment with DSM–IV criteria. BDI–II consists of 21 items, each corresponds to a symptom of depression is summed to give a single score for the BDI-II. According to that scale, the patient may have, not having or has minimal depressive symptoms if scoring: 0-13, has mild symptoms if scoring: 14-19, has moderate symptoms if scoring: 20-28 and has severe symptoms if scoring: 29-63.
Statistical analysis
Calculations were done with the statistical package SPSS, version 12.0. Data were presented as mean±SD (standard deviation) as they were normally distributed. Unpaired two-sided Student’s t test was used for comparison of means. Correlations between score of cognitive testing and demographic, clinical, diabetes-related risk factors, level of glycemic control; depression scores and NSE levels were assessed using Pearson’s test. To determine the relationship between NSE levels and cognitive function in patients with T2DM, linear regressions analyses were done using total score of cognition testing as the dependent variable as follow: as a first step, we carried out bivariate correlations between the dependent variable (scores of cognitive performance) and each of the independent variables (i.e. demographic, clinical, other risk factors, level of glycemic control; depression scores and NSE) (r and p values). Independent variables that had no significant correlations with the total score of cognitive functions testing were then excluded. The model was adjusted for confounder (as age, HbAIc and HOMA-IR). Age was controlled as convariants (entered as the first step). NSE was entered as the second step. HbA1c (Indicative of level of glycemic control) or HOMA-IR were added as the third step to ascertain whether long-term glycemic control and insulin resistance would add to the variance explained by the NSE and we then inverted steps two and three to determine the amount of variance explained by NSE after taking HbA1c or HOMA-IR into account. For all tests, values of p<0.05 were considered statistically significant.
Results
This study included 57 patients with T2DM (male = 20; females = 37), with mean age of 39.57±13.89 years and duration of illness of 7.37±5.15 years. Nearly half of the patients were uncontrolled on anti-diabetic treatment. Demographic and clinical characteristics of the studied group were shown in Table 1. Table 2 showed comparisons between patients and controls in scores of cognitive function and BDI-II. Patients had significantly lower scores of MMSE, different subsets of SBIS, WMS-R and total scores of cognitive testing (MMSE, SBIS and WMS-R) (p = 0.004) and higher scores of BDI-II (p = 0.001). In this study, nearly 75.43% (n = 43) of patients had depressive symptoms, mostly of mild/moderate type (52.63%; n = 30). Table 3 showed comparisons between patients and controls in ERPs variables.Patients had significantly prolonged latencies (p = 0.0001) and reduced amplitudes (p = 0.0001) of P300 component of ERPs. Table 4 showed comparisons between patients and controls in concentrations of glucose, HbIAc (an indicative of glycemic control), insulin and NSE. Patients had significantly higher concentrations of FBG, PBG, HbA1c, insulin and NSE. Nearly 73.68% (n = 42) of patients had higher NSE concentrations. Table 5 showed significant correlations between total scores of cognitive testing and P300 latency (r = -0.560, p = 0.010), P300 amplitude (r = 0.340, p = 0.004), age (r = -0.370, p = 0.050), duration of illness (r = -0.658, p =0.001) and concentrations of FBG (r = -0.543, p = 0.010), insulin (r = -0.365, p = 0.004) and NSE (r = -0.698, p = 0.001). BMI was positively correlated with HbA1c (r = 0.372, p = 0.021). HOMA-IR was positively correlated with insulin (r = 0.914, p = 0.001). Table 6 showed the associations between total scores of cognition, levels of NSE and HbIAc and HOMA-IR derived from linear regression analyses. We observed an association between total scores of cognitive testing and higher NSE concentrations in association with poor glycemic control and presence of IR. This relationship disappeared when we controlled for IR but persisted when we controlled for glycemic control (HbIAc).
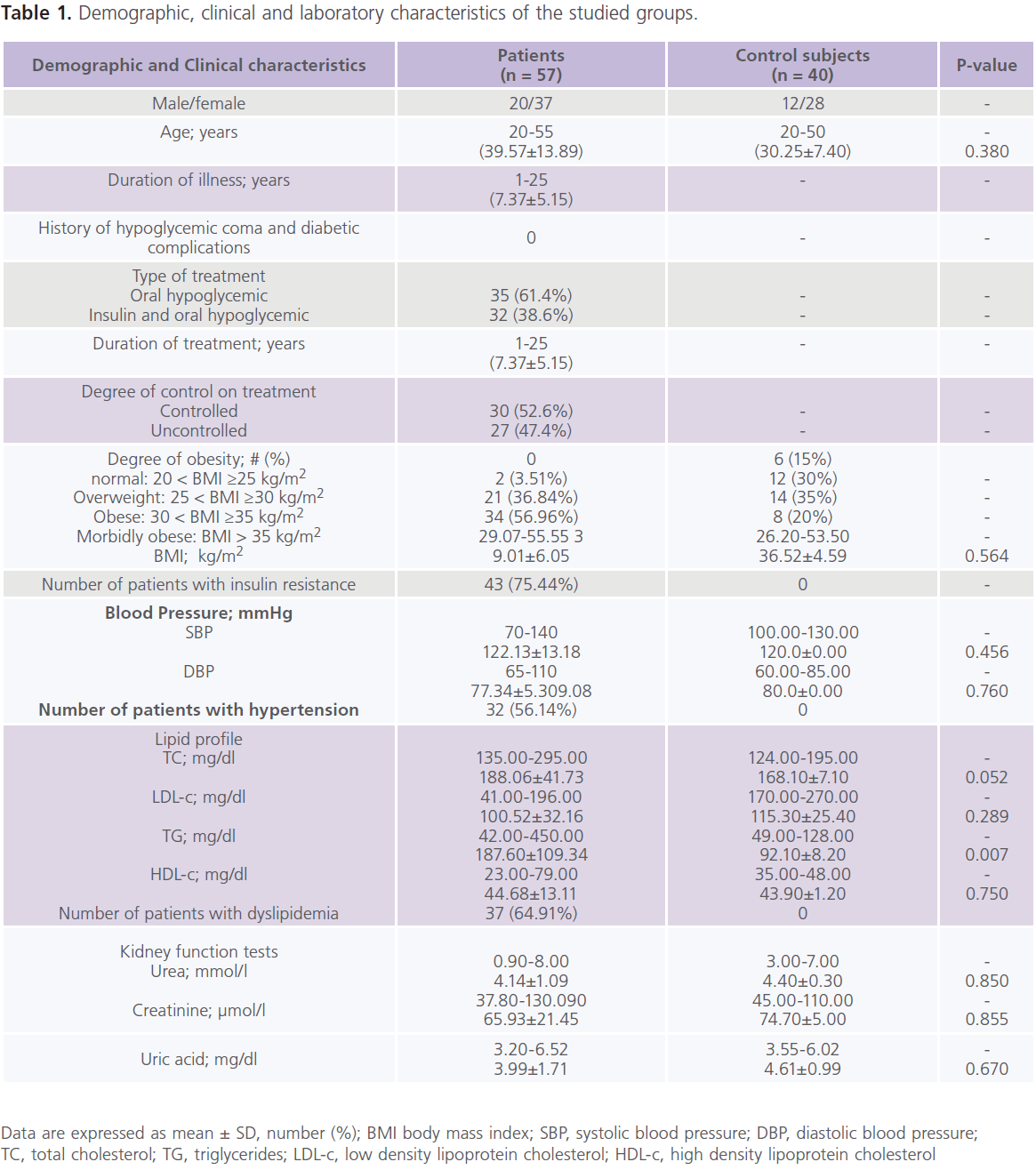
Table 1: Demographic, clinical and laboratory characteristics of the studied groups.
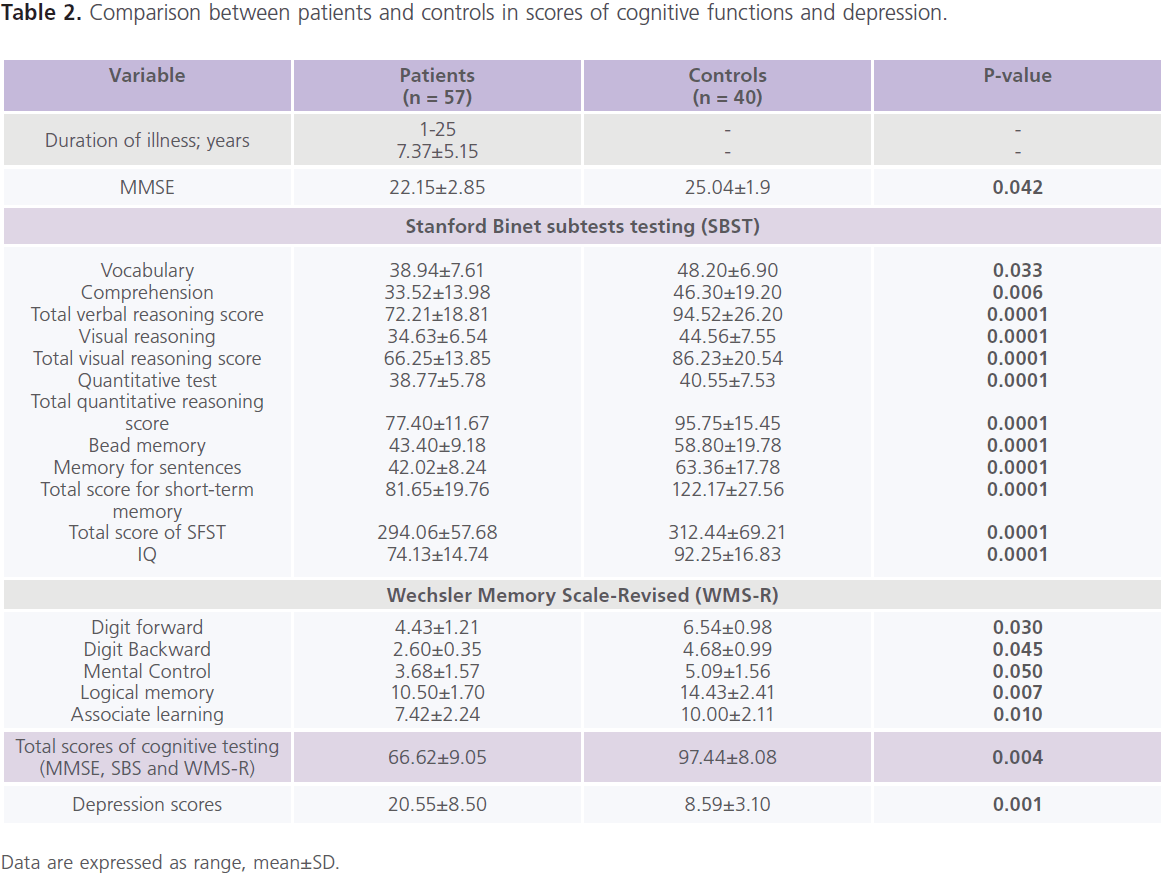
Table 2: Comparison between patients and controls in scores of cognitive functions and depression.
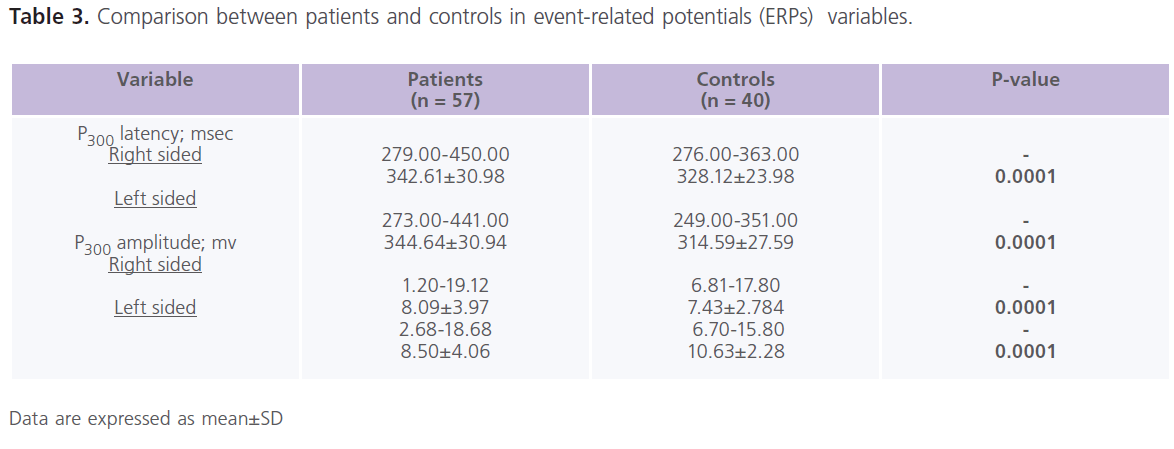
Table 3: Comparison between patients and controls in event-related potentials (ERPs) variables.
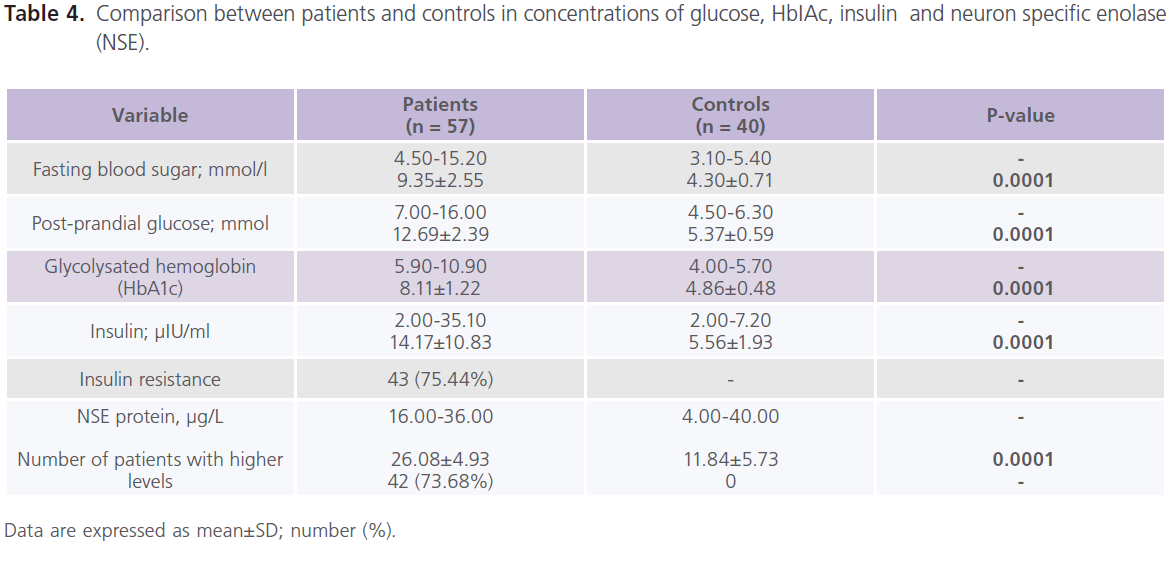
Table 4: Comparison between patients and controls in concentrations of glucose, HbIAc, insulin and neuron specific enolase (NSE).

Table 5: Pearson’s correlation (r and p-value) between total scores of cognitive testing; ERPs variables, clinical variables, lab variables, depression scores and NSE concentrations.
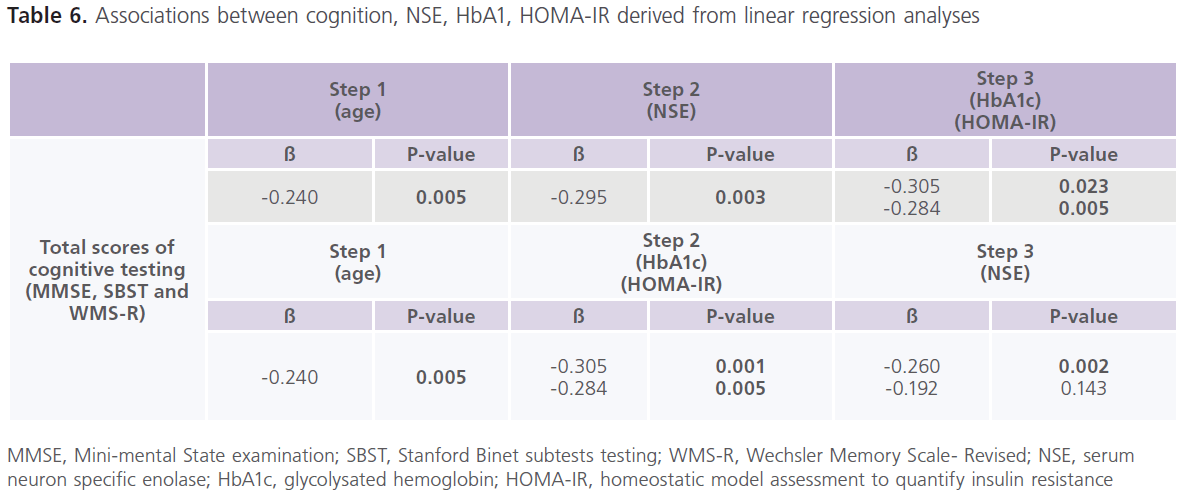
Table 6: Associations between cognition, NSE, HbA1, HOMA-IR derived from linear regression analyses
ERPs, event related potentials; MMSE, Mini-mental State examination, SBST, Stanford Binet subtests testing; WMS-R, Wechsler Memory Scale- Revised; HbA1c, glycolysated hemoglobin; SBP, systolic blood pressure; DBP, diastolic blood pressure; TC, total cholesterol; TG, triglycerides; LDL-c, low density lipoprotein cholesterol; HDL-c, high density lipoprotein cholesterol; BMI, body mass index; HOMA-IR, homeostatic model assessment to quantify insulin resistance; NSE: serum neuron specific enolase.
Discussion
Many investigators consider DM as a risk for cognitive impairment (4-8), while others reported subtle or no central nervous system (CNS) structural or functional deficits with DM (9). These controversial results are due to the fact that chronic hyperglycemia commonly develops in the context of other vascular risk factors for microvascular and macrovascular disease and atherosclerosis (3,9-16,23). However, the direct effect of chronic hyperglycemia on the brain in absence of numerous potential vascular confounders has not been fully elucidated. Thus in this study, we tried to control for numerous known potential confounders which may compromise cognition.
Evidences from this study further confirm the risk of cognitive dysfunction with T2DM (5-9,17) as follow: First: Patients with T2DM had low scores of cognitive testing and poor performance in different cognitive tasks: as verbal relations, comprehension, visual reasoning, pattern analysis, quantitation, bead memory, short-term memory and memory for sentences, digit forward, digit backward, mental control, logical memory and associate learning (44,45). Abnormalities in P300 component of ERPs, a physiological analogue of cognitive testing (9,46) also confirmed the presence of cognitive dysfunction with T2DM. Second: Despite the presence of high frequencies of patients with IR (75.44%) (3,15), HTN (56%) (12-14) and dyslipidemia (65%) (10,11), however, none of these vascular risk factors were associated with poor cognitive scores. Depression was also frequently reported (75%), but also was not associated with poor cognitive scores. In general, epidemiological studies suggested that diabetics are two to three-folds more likely to develop depression when compared to non-diabetics. In general, the prevalence of depression with DM was estimated to be 31.1% worldwide (24-26) and 32.1% in Egyptian population (47). However, the total scores of cognitive testing was found to be negatively associated with age (14,15,48-50), presence of IR as indicated by HOMA-IR (3,15), duration of T2DM and overall level of glycemic control as indicated by HbAIc. (8,50-52) In accordance, it has been observed that maintenance of good glycemic control has a small impact on cognitive function before the age of 70. Because in older adults (≥70 years), diabetes likely interacts with other dementing processes which hasten cognitive decline (as vascular and Alzheimer’s diseases) (14,15,48-50). Cox et al. (52) observed that the increase of blood glucose >15mmol/l was associated with marked decline in cognition and poor performance in arithmetic tasks. Wu et al. (50) observed that compared to treated patients, the untreated patients with DM had a 2 point decline over 2 years on MMSE with a duration of illness <5 years and a 6 point decrease on MMSE with a duration of illness ≥5 years. In addition, many authors also observed improvement in performance of cognitive testing with improvement in glucose tolerance (45,48,51).
The results of experimental, this and other studies indicate that long-standing or chronic hyperglycemia may result in brain injury with specific vulnerability to memory and learning (in which the hippocampus and related structures are their main brain-related areas). The neuroanatomical changes observed in experimental models of diabetes may accurately reflect what is occurring in the clinical setting. It is known that T2DM is characterized by hyperglycemia, IR and a relative insulin secretion defect (53). The insulin receptors are expressed in discrete neuronal populations in the CNS, including the hippocampus. Chronic hyperglycemia may impair hippocampal and amygdala structures and functions, two important structures for learning and memory processing, regardless of vascular pathology (54,55). Decreases in hippocampal insulin receptors’ activities in T2DM may contribute to behavioral deficits in type 2 rodents and cognitive deficits in humans with T2DM (56). At the experimental level, detrimental effects on learning and memory were observed in streptozotocin (STZ) rodent model of type 1 diabetes (T1DM) and genetic models of T2DM as the GKrat (57), the db/db mouse and the Zucker rat (58) as observed with Morris water maze spatial test (58,59) and inhibitory (60) or active avoidance tasks (61) and an object-discrimination task tests (7), all are indicative of impairment in hippocampus and its related structures. Experimental studies found that hippocampal long-term potentiation (LTP) was impaired, which were manifested by impairment of spatial memory and decreased expression of LTP. Third: higher concentrations of NSE protein, a marker of neuronal cell damage (28). This indicates that long-standing T2DM may result in permanent brain damage and its clinical consequences. In accordance, adults and middle aged T2DM included in this study had direct brain injury as evidenced by higher concentrations of NSE. NSE is a sensitive marker of neuronal cell damage in absence of comorbid vascular confounders and depression; and also by the significant correlation between levels of NSE and the poor performance of cognitive functions. Previous studies found that higher levels of NSE (micromolar) were associated with exacerbation of oxidative stress and neuronal apoptosis (28). Neuronal apoptosis and suppression of cell proliferation/neurogenesis were observed in the hippocampus of diabetic rodents under conditions of uncontrolled hyperglycemia (58,62-64). Furthermore, in experimental rat models, the spontaneous onset of T2DM is associated with β-amyloid and phospho-tau accumulation as well as neurite degeneration and neuronal loss [hallmarks of early Alzheimer’s disease (AD)] (65). At the clinical level, studies reported neuropsychological and memory deficits, structural brain atrophy seen in MRI brain (particularly in the limbic structures such the hippocampus and amygdala) (55,66) and deficits in hippocampal synaptic plasticity (59) in patients with T2DM (54,66) and also in patients with early manifestation of impaired glucose tolerance, however, such changes did not relate to either diabetes duration or HbA1c (66). These changes were found to be reversed with insulin replacement (59).
Insulin is a competitive inhibitor for insulin degrading enzyme (67) and thus it has been suggested that persistent elevations in insulin may interfere with peripheral Aβ clearance, and this could lead to higher Aβ concentrations in the brain (68). Another possibility is that chronic elevation of insulin concentrations in the periphery may paradoxically causes a relative hypoinsulinized state in the brain (15) and thus resultant hyperinsulinemia could actually impair cognition by disturbing insulin-mediated utilization of glucose by cells in the brain particularly the hippocampus, which is enriched with insulin receptors (69).
The knowledge that cognitive deficits are frequently associated with T2DM will has important implications for treatment of T2DM and for research purposes. However, and despite the strength of our findings, this study had some limitations which include a relatively small sample size. Future researches have to include, the following: a) longitudinal studies that prospectively assess the relation of the disease process to cognition over time, b) comprehensive longitudinal evaluation of lab markers of brain damage, cognition, and brain imaging, and c) randomized clinical trials that compare cognitive function in DM patients receiving memory enhancers, antidepressants, versus a control group of DM patients.
Conclusions
Cognitive dysfunction in T2DM appears to be due to permanent brain damage and correlated with the level of glycemic control. Large-scale epidemiological and intervention studies might enhance our understanding and management of diabetes-related cognitive and behavioral abnormalities.
Conflict of interests
The authors declare no conflict of interests. There is no involvement of sponsor for this work design, data collection, analysis, interpretation, drafting, nor the decision to submit this paper for publication. All are authors’ responsibility.
6461
References
- Bonow RO, Gheorghiade M. The diabetes epidemic: a national and global crisis, Am J Med 2004;116(suppl 5A):2S-10S.
- Dyck PJ, Kratz KM, Karnes JL, Litchy WJ, Klein R, Pach JM, Wilson DM, O’Brien PC, Melton LJ 3rd, Service FJ. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology 1993;43(4):817–824.
- Taylor VH, MacQueen GM. Cognitive dysfunction associated with metabolic syndrome. Obes Rev 2007;8(5):409-418.
- Liebson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol 1997;145(4):301-308.
- Strachan MWJ, Deary IJ, Ewing FM, Frier BM. Is type II diabetes associated with an increased risk of cognitive dysfunction? A critical review of published studies. Diabetes Care 1997;20(3):438-445.
- Stewart R, Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med 1999;16(2):93-112.
- Popoviç M, Biessels GJ, Isaacson RL, Gispen WH. Learning and memory in streptozotocin-induced diabetic rats in a novel spatial/ object discrimination task. Behav Brain Res 2001;122(2):201-207.
- Cosway R, Strachan MW, Dougall A, Frier BM, Deary IJ. Cognitive function and information processing in type 2 diabetes. Diabet Med 2001;18(10):803-810.
- Awad N, Gagnon M, Messier C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J Clin Exp Neuropsychol 2004;26(8):1044-1080.
- 10 van Exel E, de Craen AJ, Gussekloo J, Houx P, Bootsma-van der Wiel A, Macfarlane PW, Blauw GJ, Westendorp RG. Association between high-density lipoprotein and cognitive impairment in the oldest old. Ann Neurol 2002;51(6):716-721.
- Henderson VW, Guthrie JR, Dennerstein L. Serum lipids and memory in a population based cohort of middle age women. J Neurol Neurosurg Psychiatry 2003;74(11):1530-1535.
- Elias PK, Wilson PW, Elias MF, Silbershatz H, D’Agostino RB, Wolf PA, Cupples LA. NIDDM and blood pressure as risk factors for poor cognitive performance. Diabetes Care 1997;20(9):1388-1395.
- Raz N, Rodrigue KM, Acker JD. Hypertension and the brain: vulnerability of the prefrontal regions and executive functions. Behav Neurosci 2003;117(6):1169-1180.
- Hassing LB, Hofer SM, Nilsson SE, Berg S, Pedersen NL, McClearn G, Johansson B. Comorbid type 2 diabetes mellitus and hypertension exacerbates cognitive decline: evidence from a longitudinal study. Age Ageing 2004;33(4):355–361.
- Craft S. Insulin resistance syndrome and alzheimer’s disease: Ageand obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging 2005;26(supp 1):65-69.
- Draelos MT, Jacobson AM, Weinger K, Widom B, Ryan CM, Finkelstein DM, Simonson DC. Cognitive function in patients with insulindependent diabetes mellitus during hyperglycemia and hypoglycemia. Am J Med 1995;98(2):135–144.
- Ryan CM, Geckle MO. Circumscribed cognitive dysfunction in middleaged adults with type 2 diabetes. Diabetes Care 2000;23(10):1486- 1493.
- Lee ZS, Chan JC, Yeung VT, Chow CC, Lau MS, Ko GT, Li JK, Cockram CS, Critchley JA. Plasma insulin, growth hormone, cortisol, and central obesity among young Chinese type 2 diabetic patients. Diabetes Care 1999;22(9):1450–1457.
- Rosmond R. Stress induced disturbances of the HPA axis: a pathway to Type 2 diabetes? Med. Sci. Monit 2003;9(2):RA35–RA39.
- Gold SM, Dziobek I, Rogers K, Bayoumy A, McHugh PF, Convit A. Hypertension and hypothalamo-pituitary-adrenal axis hyperactivity affect frontal lobe integrity. J Clin Endocrinol Metab 2005;90(6):3262- 3267.
- Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E, Arentoft A, Wolf OT, Convit A. Hypothalamic-pituitary-adrenal axis dysregulation and memory impairments in type 2 diabetes. J Clin Endocrinol Metab 2007;92(7):2439–2445.
- Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes/Metab Res Rev 2001;17(3):189–212.
- Bekyarova GY, Ivanova DG, Madjova VH. Molecular mechanisms associating oxidative stress with endothelial dysfunction in the development of various vascular complications in diabetes mellitus. Folia Med (Plovdiv) 2007;49(3-4):13-19.
- McEwen BS, Magarinos AM, Reagan LP. Studies of hormone action in the hippocampal formation: possible relevance to depression and diabetes. J Psychosom Res 2002;53(4):883–890.
- Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications 2005;19(2):113–122.
- Verma SK, Luo N, Subramaniam M, Sum CF, Stahl D, Liow PH, Chong SA. Impact of depression on health related quality of life in patients with diabetes. Ann Acad Med Singapore 2010;39(12):913-917.
- Cooper EH, Pritchard J, Bailey CC. Serum Neuron-Specific Enolase in children’s cancer. Br J Cancer 1987;56(1):65-67.
- Skogseid IM, Nordby HK, Urdal P, Paus E, Lileaas F. Increased serum creatine kinase BB and neuron-specific enolase following head injury indicates brain damage. Acta Neurochir (Wien) 1992;115(3-4):106-111.
- Herrmann M, Ehrenreich H. Brain derived proteins as markers of acute stroke: their relation to pathophysiology, outcome prediction and neuroprotective drug monitoring. Restor Neurol Neurosci 2003;21(3- 4):177–190.
- WHO Expert Committee on Biological Standardization. Thirty-fifth report. World Health Organ Tech Rep Ser 1985;725:1-140.
- Niewoehner CB. Endocrine pathophysiology. Fence Ceek Publ., Madison, 1998.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Circulation 2002;106(25):3143-421.
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and b-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28(7):412–419.
- Paus E, Nustd K. Immunoradiometric assay for ag and gg-Enolase (neuron specific enolase), with use of monoclonal antibodies and magnetizable polymer particles. Clin Chem 1989;35(10):2034-2038.
- Folstein MF, Folstein SE, McHugh PH. “Mini-mental state” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189-198.
- Al-Rajeh S, Ogunniyi A, Awada A, Daif A, Zaidan R. Preliminary assessmet of an Arabic version of the mini-mental state examination.. Annals of Saudi Medicine 1999;19(2):150-152.
- Delany EA, and Hopkins TF. The Stanford. Binet Intelligence scale: fourth Edition: Examiner‘s Handbook. Chicago. The Riverside Publishing Co, 1986.
- Melika LK. The Stanford. Binet Intelligence Scale, fourth edition. Arabic Examiner’s Handbook. Cairo: Dar El Maref Publishing; Egypt, Cairo, 1998.
- Wechsler D. Wechsler Memory Scales-Revised. New York: Psychological cooperation, 1987.
- Polich J. P300 clinical utility and control of variability. J Clin Neurophysiol 1998;15(1):14–33.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (4th ed.) Washington, DC: American Psychiatric Association, 1994;317-391.
- Gharyb AG. Beck Depression Inventory–II (BDI-II), Arabic Examiner’s Handbook. Dar El -Anglo Publishing, Egypt, Cairo, 2000.
- Beck AT, Steer RA, Ball R, Ranieri W. Comparison of Beck Depression Inventories -IA and -II in psychiatric outpatients”. Journal of personality assessment 1996;67(3):588–597.
- Mooradian AD, Perryman K, Fitten J, Kavonian GD, Morley JE. Cortical function in elderly non-insulin dependent diabetic patients: behavioural and electrophysiological studies. Arch InternMed 1988;148(11):2369-2372.
- Gradman TJ, Laws A, Thompson LW, Reaven GM. Verbal learning and/or memory improves with glycemic control in older subjects with non-insulin-dependent diabetes mellitus. J Am Geriatr Soc 1993;41(12):1305-1312.
- Kurita A, Mochio S, Isogai Y. Changes in auditory P300 event related potentials and brainstem evoked potentials in diabetes mellitus. Acta Neurol Scand 1995;92(4):319-323.
- Shehatah A, Rabie MA, Al-Shahry A. Prevalence and correlates of depressive disorders in elderly with type 2 diabetes in primary health care settings. J Affect Disord 2010;123(1-3):197-201.
- Naor M, Steingrüber HJ, Westhoff K, Schottenfeld-Naor Y, Gries AF. Cognitive function in elderly non-insulin-dependent diabetic patients before and after inpatient treatment for metabolic control. J Diabetes Complications 1997;11(1):40-46.
- Convit, A, Wolf OT, Tarshish C, de Leon MJ. Reduced glucose tolerance is associated with poor memory performance and hippocampal atrophy among normal elderly. Proc Natl Acad Sci USA 2003;100(4):2019-2022.
- Wu JH, Haan MN, Liang J, Ghosh D, Gonzalez HM, Herman WH. Impact of diabetes on cognitive function among older Latinos: a population-based cohort study. J Clin Epidemiol 2003;56(7):686-693.
- Meneilly GS, Cheung E, Tessier D, Yakura C, Tuokko H. The effect of improved glycemic control on cognitive functions in the elderly patient with diabetes. J Gerontol 1993;48(4):M117-121.
- Cox DJ, Kovatchev BP, Gonder-Frederick LA, Summers KH, McCall A, Grimm KJ, Clarke WL. Relationships between hyperglycemia and cognitive performance among adults with type 1 and type 2 diabetes. Diabetes Care 2005;28(1):71-77.
- Kasuga M. Insulin resistance and pancreatic beta cell failure. J Clin Invest 2006;116(7):1756-1760.
- Gold AE, Deary IJ, Jones RW, O’Hare JP, Reckless JPD, Frier BM. Severe deterioration in cognitive function and personality in five patients with long-standing diabetes: a complication of diabetes or a consequence of treatment? Diabet Med 1994;11(5):499–505.
- den Heijer T, Vermeer SE, van Dijk EJ, Prins ND, Koudstaal PJ, Hofman A, Breteler MM. Type 2 diabetes and atrophy of the medial temporal lobe structures. Diabetologia 2005;46(12):1604-1610.
- Park CR. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev 2001;25(4):311-323.
- Marfaing-Jallat P, Portha B, Penicaud L. Altered conditioned taste aversion and glucose utilization in related brain nuclei of diabetic GK rats. Brain Res Bull 1995;37(6):639–643.
- Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor deficient rodents. Neuroscience 2002;113(3):607–615.
- Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res 1998;800(1):125–135.
- Baydas G, Nedzvetskii VS, Nerush PA, Kirichenko SV, Yoldas T. Altered expression of NCAM in hippocampus and cortex may underlie memory and learning deficits in rats with streptozotocin-induced diabetes mellitus. Life Sci 2003;73(15):1907–1916.
- Flood JF, Mooradian AD, Morley JE. Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes 1990;39(11):1391–1398.
- Kim HB, Jang MH, Shin MC, Lim BV, Kim YP, Kim KJ, Kim EH, Kim CJ. Treadmill exercise increases cell proliferation in dentate gyrus of rats with streptozotocin-induced diabetes. J Diabetes Complications 2003;17(1):29–33
- Beauquis J, Roig P, Homo-Delarche F, De Nicola A, Saravia F. Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: reversion by antidepressant treatment. Eur J Neurosci 2006;23(6):1539–1546.
- Saravia FE, Beauquis J, Revsin Y, Homo-Delarche F, de Kloet ER, De Nicola AF. Hippocampal neuropathology of diabetes mellitus is relieved by estrogen treatment. Cell Mol Neurobiol 2006;26(4-6):943– 957.
- Manning CA, Ragozzino ME, Gold PE. Glucose enhancement of memory in patients with probable senile dementia of the Alzheimer’s type. Neurobiol Aging 1993;14(6):523–528.
- Manschot SM, Brands AM, van der GJ, Kessels RP, Algra A, Kappelle LJ, Biessels GJ. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 2006;55(4):1106–1113.
- Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: Progress and potential. Endocr Rev 1998;19(5):608-624.
- Fishel MA, Watson GS, Montine TJ, Wang Q, Green PS, Kulstad JJ, Cook DG, Peskind ER, Baker LD, Goldgaber D, Nie W, Asthana S, Plymate SR, Schwartz MW, Craft S. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch Neurol 2005;62(10):1539-1544.
- Zhao WQ, Alkon DL. Role of insulin and insulin receptor in learning and memory. Molecular and Cellular Endocrinology 2001;177(1-2):125- 134.

