Keywords
Orlistat, cholesterol, particle size.
Introduction
Drug delivery system using colloidal particulate carrier such as liposomes [1] or niosomes [2] have distinct advantages over conventional dosage forms because the particles can act as a drug containing reservoirs. Modification of the particle composition or surface can adjust the affinity for the target site and / or the drug release rate. The slowing drug release rate may reduce the toxicity of drug so these carriers play an increasingly important role in drug delivery. Niosomes are non-ionic surfactant based vehicles that had been developed as alternative controlled drug delivery systems to liposomes may be unilamellar or multilamellar depending on the method used to prepare them and they have been extensively studied for their potential to serve as carriers for delivery for drugs, antigens, hormone and other biogenetic agents. The ultimate aim in developing delivery system is controlling the release of drugs from the carrier system, in order to achieve an extended uptake in the body.
Niosomes similar to liposomes are biodegradable, biocompatible and non-imunogenic in nature and exhibit flexibility in their structured characterization [3]. In addition needlessness of handling or storing of niosomes in special conditions [2, 4] and the availability as well as in expensiveness of prepared materials. Even though niosomes exhibit good chemical stability during storage there may be problems of physical stability like aggregation, fusion and leaking etc. The additional convenience of the transportation, distribution, storage and dosing would make ‘dry niosomes’ a promising industrial product [5, 6]. This dry, free flowing granular product which, upon addition of water, disperses or dissolves to form a multilamellar niosomes suspension suitable for administration by oral or other routes.
Orlistat (tetrahydrolipstatin) is the first agent of novel noncentrally acting anti-obesity agent that acts locally in the gastrointestinal tract to inhibit pancreatic and gastric lipases, derived from lipstatin, a natural product of Streptomyces toxytricin [7]. By covalently blocking the lipase active site, Orlistat inhibits the hydrolysis of dietary Triglycerides (TGs) and thus reduces the subsequent intestinal absorption of the lipolysis products monoglycerides (MGs) and free fatty acids (FFAs). At the recommended therapeutic dose of 120 mg three times a day, Orlistat inhibits dietary fat absorption by about 30% [8]
The aim of this study is to investigate the feasibility of using proniosomes as stable precursors for the preparation of niosomes as drug carriers systems for poorly- soluble drugs. Proniosomes provide the advantages of easy and immediate preparation of niosomes and also enhancing the solubility of poorly soluble drugs, controlling its release and prolonging its activity over long periods of time. Hence decreasing the frequency of administration and improving patient compliance. The influence of different processing and formulation variables such as cholesterol content and non ionic surfactant ratio, drug concentration, carrier and the pH of the hydration medium on orlistat entrapment efficiency will be demonstrated. Also, orlistat release rates from proniosomes in 0.1 N HCL buffer, its morphology and release kinetics will be illustrated.
Materials and Methods
Orlistat is a gift sample from Glukem Pharma, Hyderabad, Cholesterol and Cyclodextrin were obtained from S.d fine Chem limited, Mumbai, Span 60 Loba Chemie Pvt ltd, Mumbai and all the other chemicals were of analytical grade.
Preparation of Niosomes from Proniosomes:
Preparation of niosomes from proniosomes by hydrating the obtained proniosomes with 0.5% NaCl containing 3% SLS at pH 6.0 buffer by gentle mixing in a round bottom flask. The niosomes were sonicated twice for 30min using probe sonicator (Vibronics, Mumbai) and then freeze dried, preserved for further studies.
Preparation of Proniosomes:
For the preparation of 250μmol stock solution of surfactant and cholesterol was dissolved in Chloroform: Methanol (2:1). Different niosomal preparations (OT1, OT2, OT3, OT4, OT5, OT6, OT7, OT8, OT9) having different proportions of Span60 and cholesterol (i.e, 0.9:0.1, 0.8:0.2, 0.7:0.3, 0.6:0.4, 0.5:0.5, 0.4:0.6, 0.3:0.7; 0.2:0.8, 0.1:0.9) were prepared by mixing both span60 and cholesterol dissolved in chloroform: methanol (2:1) and Orlistat (25mg) dissolved in solvent system (chloroform: methanol (2:1)) the drug solution was added to 100ml round bottom flask containing the span60 and cholesterol stock solution with 1gm of Cyclodextrin. Additional organic solution added to form slurry. The flask was attached to a rotary flash evaporator (Superfit, Mumbai) to evaporate solvent by rotating at 60 to 70 rotations per minute (rpm), a temperature of 45 ± 20C and a reduced pressure of 600mmHg until the mass in the flask had become a dry, free flowing product.
In –vitro characterization of Niosomes Surface Morphology
The surface morphology of the prepared niosomes was examined under by using Scanning electron microscope (SEM) (Jeol, JSM-6360, Japan). The niosomal formulation were subjected to freeze drying then resulting solid content was mounted on to screw shaped stubs using double – sided carbon adhesive tape [9, 10]. The samples were coated with platinum in an argon atmosphere under vacuum condition by using ion sputter chamber and they were examined at 15000v accelerating voltage.
Particle Size
Particle size of the prepared niosomal formulations were determined by particle size analyzer (Microtract S3500, USA). 1ml of the niosomal samples was poured in to the sampling hollow chamber and the mean particle size was detected using software system.
Drug Content
The drug content of the formulated niosomal formulations was determined by HPLC [11]. Isocratic C13 was used as a column using 0.1% phosphoric acid and acetonitrile (1: 9) as a solvent system at a flow rate of 0.7ml / min. Sampling running time was maintained for every 20 minutes interval. Percentage (%) Drug content was calculated by using the standard surface area as well as sample surface area multiplied by 100.

In- Vitro Drug Release
The in- vitro drug release of niosomal formulations was performed by using dialysis method [12] in an open ended tube sealed with dialysis membrane ( Himedia lab pvt ltd, Mumbai, India) which was fitted in a USP dissolution apparatus containing 200ml of 0.1N HCl as dissolution medium stirred at 75 rpm of temperature 37°C ± 0.5°C. Niosomal Formulation (25ml) was added in to the dialysis tube and samples (5ml) were with drawn at predetermined time intervals for a period of 12 hrs and replaced by the same amount of fresh buffer to maintain sink condition. Absorbances of withdrawn samples were measured using UV- Visible spectrophotometer at 254 nm. The amount of drug release was obtained from the standard calibration curve. The data obtained from in- vitro drug release were fitted with various kinetic equations like Zero order, first order, Higuchi, Korsmeyer- Pappas and Hixson Crowell equation.
Fourier Transform Infrared Analysis (FT-IR)
Fourier Transform Infrared Analysis (FT-IR) measurements [13] of pure drug, carrier and drugloaded niosomal formulations were obtained using a Perkin- Elmer system 200 FT-IR spectrophotometer. The pellets were prepared on KBr-press under hydraulic pressure of 150kg / cm2; the spectra were scanned over the wave number range of 4000 to 400 cm-1 at the ambient temperature.
Density
Weigh the empty, dry pycnometer as (W1), fill the pycnometer with distilled water up to its neck and measure the weight as (W2). Then fill the pycnometer with the formulation and measure the weight as (W3) then calculate the density 14 of the liquid formulation by using the formula.
Density of water = W2-W1 / Volume
Density of sample = W3-W2/ W2-W1×Density of water
Viscosity
Viscosity determination was done by using Ostwald viscometer. The niosomal formulation was poured in to the apparatus through the left arm up to the mark A. The formulation was sucked in to the right arm slightly above the point B and the left arm was closed with the thumb to keep the liquid with out dropping down. The apparatus was clamped vertically and the thumb was removed so as to allow the liquid to fall through the capillary under gravity then note down the time taken for the formulation to drop down from the point B to C. Then calculate the viscosity 14 by using poisulles equation as.
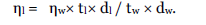
pH
pH of the niosomal formulations were measured using digital pH meter by placing the pH meter in a glass beaker containing formulated preparations. The pH meter directly reads out the pH of the formulation.
Conductivity
Conductivity of the niosomal formulations were done by using conductivity meter 304 (Systronics, India). Initially calibrate the instruments by using 0.1N KCl and adjust the instrument to read 100. Then placed the prepared formulation and note down the conductivity.
Sedimentation Rate
The Sedimentation of suspended particle of prepared niosomes were determined by measuring the changes in nephloturbidimetric units using a digital nephloturbidity [15] meter (Model -32 M/S Systronics, India) at regular time intervals for a period of 12hrs.
Stability studies
The formulated niosomal formulations of OT1, OT3, OT5, OT7 and OT9 were tested for its stability. All formulations divided in to three sets and were stored at 4° ± 2°C, 45°± 2°C and 37°± 2°C for 3months. After 3 months, the drug content as well as the particle size was determined by the method discussed previously.
Solubility Studies of pure drug
Weighed accurately about 10gm of pure drug and dissolved each in 5ml of the solvent system i.e. water, chloroform, toluene, hexane, ethanol, nbutanol, methanol, acetone in a well closed air tight containers. Then added the successive amount of the drug in to the containers containing solvent until the solution became saturated solution. Then the containers were placed in a thermostat shaker for 24hrs and then calculate the percentage solubility [14] in each container.
Partition Co-efficient Of Pure Drug
Accurately weighed amount of 200mg drug was dissolved in one of the phases, is shaken with the other partitioning solvents such as n-butanol, chloroform, diethyl ether etc for 30min, allowed to stand for 5 minutes, and then extract the lower and upper portions separately. Then the partitioning coefficient [14] was calculated by using the formula as follows.
KWo = Concentration of drug in organic portion / Concentration of drug in aqueous phase.
Measurement of Angle of Repose
The angle of repose of dry proniosome powder was measured by a funnel method [16]. Briefly, the pure drug and proniosome powder was poured into a funnel which was fixed at a position so that the 13mm outlet orifice of the funnel is 10cm above a level black surface. The powder flowed down from the funnel to form a cone on the surface and the angle of repose was then calculated by measuring the height of the cone and the diameter of its base.
Excipient Compatibility studies
Weighed accurately about 100mg each of powder drug, cholesterol and Cyclodextrin. Then admix drug and cholesterol (1:1), drug and Cyclodextrin (1:1), cholesterol and Cyclodextrin (1:1) in an air tight screw cap amber colored vials. Individual drug, cholesterol, Cyclodextrin also placed in air tight screw cap amber colored vials, then kept the vials at room temperature as well as in hot air oven at 400C for one week 14 and carry out FT-IR analysis with saturated potassium bromide using pellet making method.
Results
Surface Morphology
Shape and surface characteristic of niosomes were examined by Scanning Electronic Microscopy analyses were summarized in (Fig. 1A to Fig. 1E). Scanning electron microscopy of best formulation OT9 shows the smooth surface with an average mean particle size of 100nm. Surface morphology illustrates the smooth surface of niosomal formulation. The prepared vesicles were studied under various magnifications of 1000X, 10,000X, 20,000X, 30,000X and 50,000 x to observe the formation of vesicles. Some unevenness of vesicles that observed under the study may be due to drying process under normal environment condition. The particles found to be uniform in size and shape.

Fig. 1. (A) SEM image of OT9 formulation at 1,000 X magnification (B) at 10000 X magnification (C) at 20000 X magnification (D) at 30000 X magnification (E) at 50000 X magnification.
Particle Size
The mean particle size of the pure Orlistat using sieving method was found to be 4.6678μ. And the particle size analysis of the formulated niosomal preparations shows the average size of <600 nm as shown in Table 1. It was clear that, the formulation OT9 mean particle size was smaller (381 nm) than all other formulations was summarized in (Fig.2A to Fig. 2E). This is because of the fact, as cholesterol increases the chain order, stabilizes the bilayers of vesicles, especially smaller ones. It was expected that vesicles with relatively high cholesterol content be smaller than the vesicles with low amounts of cholesterol. Sonication may be responsible for the breakdown of the multilamellar vesicles to form unilamellar ones [17]. The use of high cholesterol content in the formulation of Orlistat niosomes may lead to smaller vesicles. This finding may be owing to the influence of certain preparation conditions such as the hydration time and the degree of shaking.
| Formulation Code |
Surfactant:
Cholesterol (µmol) |
Particle Size (nm) |
| OT1 |
0.9 : 0.1 |
1614 ± 1615* |
| OT3 |
0.7 : 0.3 |
783 ± 773* |
| OT5 |
0.5 : 0.5 |
579 ± 571* |
| OT7 |
0.3 : 0.7 |
483 ± 485* |
| OT9 |
0.1 : 0.9 |
381 ± 363 |
Table 1: The composition and Particle Size profiles for OT1, OT3, OT5, OT7 and OT9 formulations.
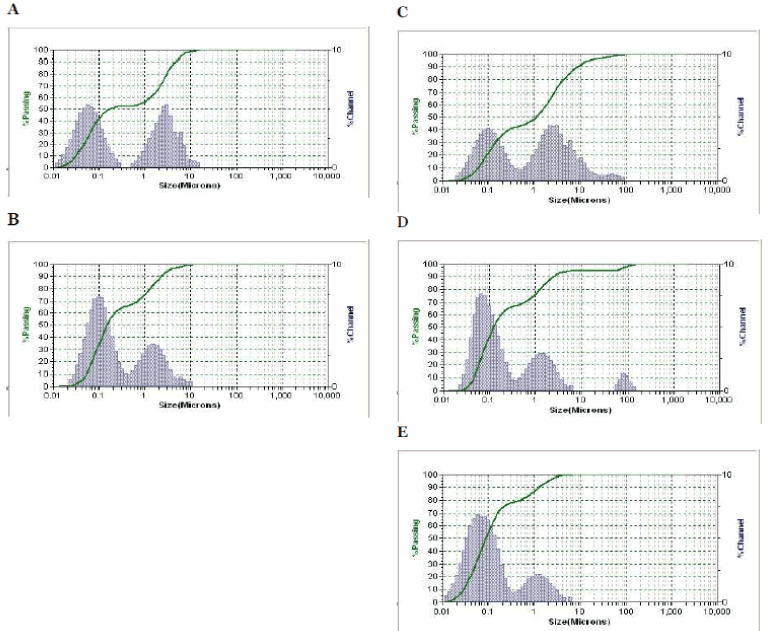
Fig. 2. Particle size for (A) OT1 (B) OT3 (C) OT5 (D) OT7 (E) OT9.
Entrapment efficiency or drug content was determined by HPLC and was expressed as a percentage of the total amount of Orlistat used initially. The calculated average percentage drug content of the niosomes was obtained from the HPLC spectrum as shown in the (Fig. 3A to Fig. 3E) by observing the surface area of sample with that of the pure drug were summarized in Table 3 (37.31 ± 10.27 %) (n = 3). Among all the formulations OT9 exhibits higher drug content (55.90). This result may be explained by the high cholesterol content and the use of less span 60 with its high phase transition temperature (TC). Entrapment of drug was increased with increasing cholesterol content when niosomes were prepared by changing the molar ratio of nonionic surfactant to cholesterol [17]. In fact, the vesicles prepared with span 60 showed the most drug content because of its highest phase transition temperature of ≈ 50°C.
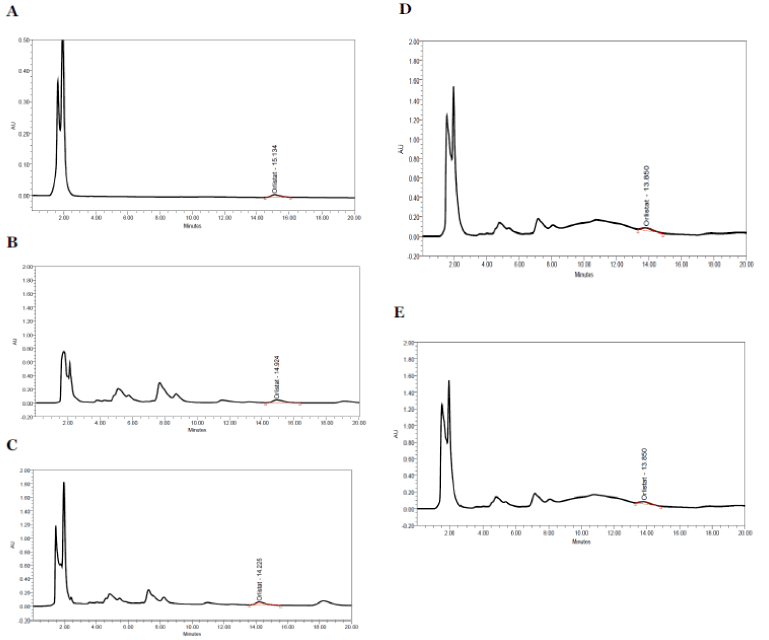
Fig. 3. High performance liquid chromatography (HPLC) spectrum of (A) Orlistat (B) OT3 (C) OT5 (D) OT7 (E) OT9.
In- Vitro Drug Release
The release study was conducted for all the formulations as shown in the Fig. 4A. Most of the formulations were found to have a linear release and the formulations were found to provide approximately 95% release with in 4hrs. It shows that there is increase in solubility which may be due to the presence of surfactants and β- cyclodextrin. The formulations which have high cholesterol ratio (OT9) were found to sustained the drug release. Cholesterol, which has a property to abolish the gel to liquid transition of niosomes, this prevents the leakage of drug from the niosomal formulation. The slower release of drug from multilamellar vesicles may be attributed to the fact that a multilamellar vesicle consists of several concentric sphere of bilayer separated by aqueous compartment. The above specified formulation OT1, OT5 and OT7 were found to give a cumulative release of 95% over a period of 4hrs, the higher release from formulation OT1 may be because of its low cholesterol content and higher surfactant levels. Formulation OT9 having the highest cholesterol content showed the sustained release over 12hrs, which gives a cumulative release of 94.59% as shown in Fig. 4B. The in-vitro release data was applied to various kinetic models to predict the drug release kinetic mechanism. The release constant was calculated from the slope of appropriate plots and the regression co-efficient (r2) was determined. It was found that the in-vitro drug release of niosomes was best explained by Hixson Plot kinetics for best formulation OT9 as the plot shows highest linearity as shown in Fig. 4C. The correlation coefficient (r2) was found 0.9659 for OT9.
Fig. 4. (A) Comparative in- vitro drug release profile of OT1, OT5, OT7 and OT9 formulations (B) Comparative in- vitro drug release profile for OT9 formulation (C) Hixson Plot kinetics for best formulation OT9.
Fourier Transform Infrared Analysis (FT-IR) of OT9
FT-IR spectra of pure Orlistat and OT9 formulation were recorded. The orlistat present in the formulation OT9 was confirmed by FT-IR. No predominant drug interaction was detected as shown in Fig. 5A and B. Although there were some mild shift in the wave number 3600- 3200 cm-1 and 1500- 8000 cm-1. The region 3600- 3200 cm-1 was a stretching region of the functional group N-H, C-H of aromatic ring (3100- 3000 cm-1), O-H (3200 cm-1) and C-H of alkenes (3100- 3000 cm-1) and C-H of alkane (3000 cm-1). The region 1500- 800 cm-1, the weak drug and polymer bound formation was noticed at 1154.61. All these peaks have appeared in pure orlistat and formulation OT9 indicating no chemical interaction between orlistat and excipients and presence of drug. It also confirmed that the stability of drug during formulation.
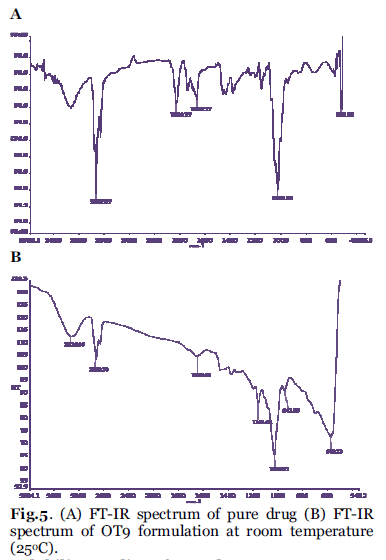
Fig. 5. (A) FT-IR spectrum of pure drug (B) FT-IR spectrum of OT9 formulation at room temperature (25°C).
Solubility Studies of pure drug
Results of the measurement of solubility of pure Orlistat are summarized in Table 2 and it indicate that the pure drug orlistat was freely soluble in alchoholic solvents such as methanol, ethanol and nbutanol and in chloroform when compared to the acetone, n-hexane, toluene and also it was observed that the pure orlistat was poorly soluble in water. The vegetable oils such as PEG-400, PEG-600, glycerol and propylene glycol reveal that it may be sparingly soluble.
| S.No |
Solvent system (ml) |
Solubility (gm/ml) |
| 1 |
Water |
0.001 |
| 2 |
Chloroform |
0.750 |
| 3 |
Toluene |
0.600 |
| 4 |
Hexane |
0.550 |
| 5 |
Acetone |
0.750 |
| 6 |
Ethanol |
1.300 |
| 7 |
n-Butanol |
1.250 |
| 8 |
Methanol |
1.500 |
| 9 |
PEG 400 |
0.060 |
| 10 |
PEG 600 |
0.060 |
| 11 |
Glycerol |
0.075 |
| 12 |
Propylene glycol |
0.060 |
| 13 |
Sesame oil |
0.050 |
| 14 |
Olive oil |
0.050 |
| 15 |
Castor oil |
0.050 |
Table 2: Solubility Studies Data of Pure Drug
Partition Co- Efficiency of Pure Drug
Partition co- efficiency of pure drug was determined using various organic phases along with aqueous phase were summarized in Table 3. Results revealed that the pure drug orlistat exhibits highly liphophillic in nature.
| S.No |
Solvent system (1:1) |
Partition Co-efficient |
| 1 |
Water: n-Butanol |
9.2880 |
| 2 |
Water :Diethyl ether |
4.3785 |
| 3 |
Water: Chloroform |
-0.5496 |
| 4 |
Water: Di isopropyl |
67.6880 |
| 5 |
Water: Benzene |
0.0626 |
| 6 |
Water: n-heptane |
0.7147 |
| 7 |
Water: Cyclohexane |
6.0137 |
Table 3: Partition Co-efficient of drug using different solvents
Measurement of angle of repose
The angle of repose of pure drug and dry proniosome powder formulations of OT5, OT7 and OT9 were measured out using funnel method. The free flowing property of pure drug was very poor when compared with the prepared formulations of OT5 (10.58 ±0.0124), OT7 (19.42 ± 0.0001) and OT9 (16.86 ± 0.0001). This was due to the fact that the pure drug was sticking to the edges of the funnel because of its sensitive nature towards moisture. The free flowing results in the prepared niosomal formulations were due to the addition of carrier (β-CD) as well increase in size of particle with excipients (span 60 and cholesterol). In fact, if the proportion of cholesterol to surfactant in the formulation increases the flow properties of dry proniosome powder decreased.
Excipient compatibility studies
FT-IR studies were done to confirm and detect drug presence and possible interaction between the Orlistat and excipients that are used in the formulation. Fig. 6A, B and C shows the IR spectrum of the drug Orlistat with physical mixture of cholesterol cyclodextrin (1:1), and physical mixture of cholesterol and cyclodextrin (1:1) respectively. Results revealed that there was slight shift in the band range of 3340.94, 3340.93 and 3399.20 in Fig. 6A, B and C respectively. When compared with pure spectrum band of 3339.32 this may be due to the bonding between the β-CD and the Orlistat. And the other bands in the spectrum 2955.18, 2918.20, 2955.18 shows that the drug was not degraded (or) decomposed, it confirms that the drug present in the physical mixtures as shown in Table 4. It was supported with drug content.
| S. No |
Ingredients |
Inference |
| 1 |
Drug |
(-) |
| 2 |
Span60 |
(-) |
| 3 |
Cholesterol |
(-) |
| 4 |
Cyclodextrin |
(-) |
| 5 |
Drug: cholesterol |
(-) |
| 6 |
Drug: Cyclodextrin |
(-) |
| 7 |
Cholesterol: Cyclodextrin |
(-) |
(+) indicates interaction, (-) indicates no interaction.
Sedimentation Rate .
Table 4.Excipient Compatibility Studies for pure drug Orlistat.
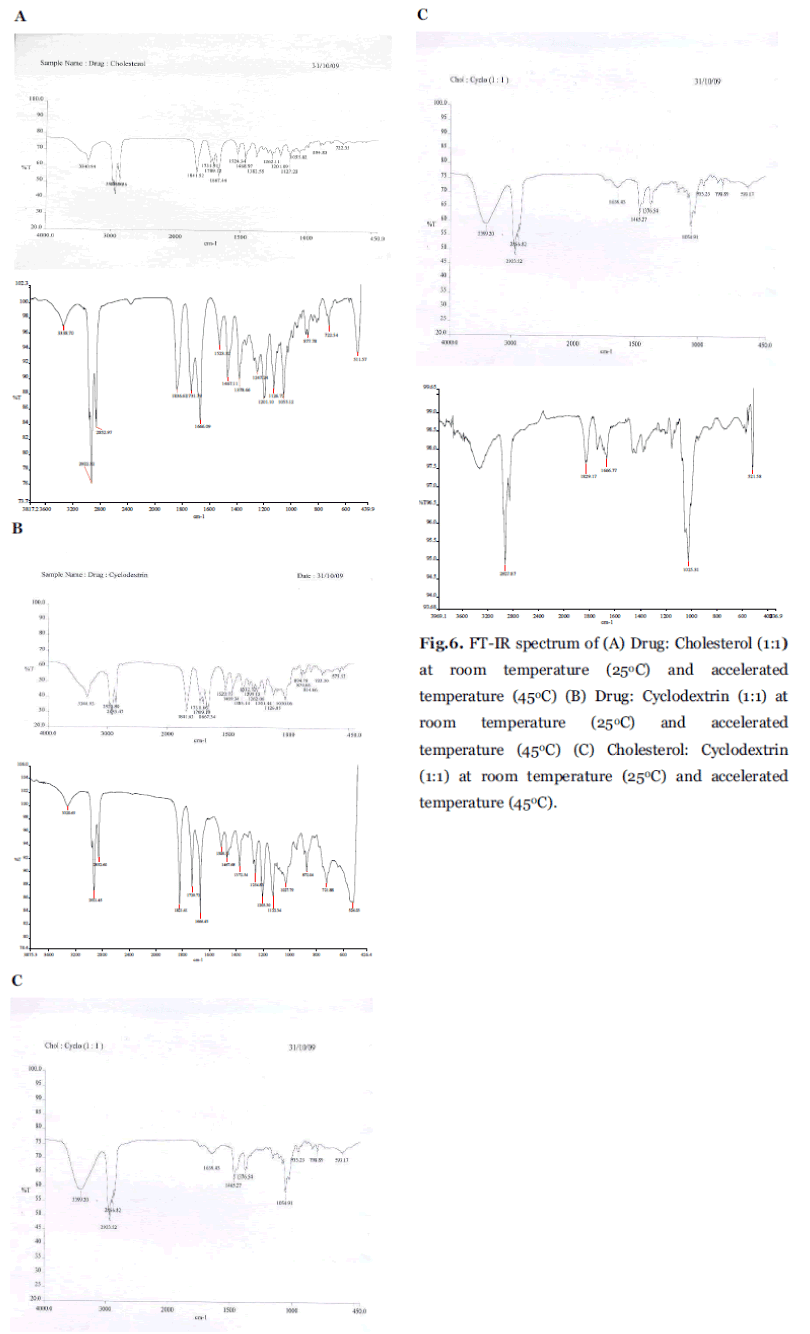
Fig. 6. FT-IR spectrum of (A) Drug: Cholesterol (1:1) at room temperature (25°C) and accelerated temperature (45°C) (B) Drug: Cyclodextrin (1:1) at room temperature (25°C) and accelerated temperature (45°C) (C) Cholesterol: Cyclodextrin (1:1) at room temperature (25°C) and accelerated temperature (45°C).
Sedimentation Rate
Sedimentation rate was determined by using nephloturbidimeter and the result was obtained by plotting a graph between times versus N.T.U unit. The sedimentation rate for the formulation OT9 was shown in Fig. 7. It was clear that, there was no sedimentation of the niosomes even after standing for 12hrs at room temperature. It shows the stability of the formulation.

Fig. 7. FT-IR spectrum of (A) Drug: Cholesterol (1:1) at room temperature (25°C) and accelerated temperature (45°C) (B) Drug: Cyclodextrin (1:1) at room temperature (25°C) and accelerated temperature (45°C) (C) Cholesterol: Cyclodextrin (1:1) at room temperature (25°C) and accelerated temperature (45°C).
Density, pH, viscosity and conductivity
Density, pH, viscosity and conductivity was measured out for all formulations as shown in Table 5 and it shows that viscosity of the OT9 formulation is less when compared to all other formulations due to the presence of low surfactant level. Density of OT9 is more due to the high cholesterol content.
| Formulation Code |
Surfactant: Cholesterol (µmol) |
Density (gm/ cm3) |
Viscosity (µpas) |
pH |
Conductivity (µ?) |
| OT1 |
0.9 : 0.1 |
1.0498 ± 0.002* |
2.0678 × 10-3 |
7.6 |
6.88 ± 0.008* |
| OT3 |
0.7 : 0.3 |
1.0293 ± 0.0001* |
2.0637 × 10-3 |
7.4 |
7.40 ± 0.008* |
| OT5 |
0.5 : 0.5 |
1.0773 ± 0.0001* |
1.5289 × 10-3 |
7.8 |
8.02 ± 0.056* |
| OT7 |
0.3 : 0.7 |
1.0543 ± 0.0001* |
2.0822 × 10-3 |
8.0 |
|
| OT9 |
0.1 : 0.9 |
1.0466 ± 0.0001* |
2.1523 × 10-3 |
6.0 |
7.53 ± 0.012* |
| 0.008* |
|
|
|
|
7.43 ± |
* Standard Deviation
Stability studies.
Table 5:Density, Viscosity, pH and conductivity profiles for OT1, OT3, OT5, OT7 and OT9 formulations.
Stability studies
The niosomes have nature of protecting the components from light and moisture though the Orlistat is both light and moisture sensitive it was stable through out the period of stability study it was confirmed by drug content. After 3 months, the mean particle size of the OT9 formulation shows slight change in mean particle size upon storage at 37° ± 2°C (134 nm), 4° ± 2°C (225nm) and 45° ± 2°C (446nm). This suggests that niosomes offered more stable system that could minimize the problems associated with conventional dosage forms.
Conclusion
The results of this study show that non-ionic surfactants can be used for the preparation of orlistat entrapping niosomes. The surfactant level, the cholesterol content and charge inclusion altered the entrapment efficiency, size distribution range and drug release rate from niosomes. Niosomes composed of span 60 and cholesterol was found to most effectively sustain the release of poorly water soluble drug Orlistat. Thus niosomes acts as novel carriers in order to improving solubility by addition of a carrier like β- CD. Enchanced absorption leads to enhance bioavailability of Orlistat. To protect from light and degradation the drug were encapsulating in the form of niosomes there by improving stability of the formulations. Finally to facilitate sustain release of orlistat. Thus niosomes acts as promising sustained drug delivery carriers in the formulation of poorly water soluble drugs like Orlistat.
Acknowledgement
The authors are gratefully acknowledged the SASTRA University for providing necessary facilities and support and financial assistance from TNSCST, Tamil Nadu, India; the gift sample of Orlistat supplied by Glukem pharmaceuticals (Hyderabad).
Conflict of Interest: NIL
Source of Support: NONE
5659
References
- M. Reza mozafari, Bioactive entrapment and targeting using nanocarrier technologies: An introduction Frontiers of nanotherapy. University of Michigan, Springer, (2006)1-6.
- Varaporn Buraphacheep Junyaprasert, Veerawat Teeranachaideekul, Tasaneey Supaperm, Effect of charged and non-ionic membrane additives on physicochemical properities and stability of niosomes, AAPS Pharn sci tech. 9 (2008) 851-59.
- Nefise Ozlen Sahin, Niosomes as new carrier systems, Nanomaterials and nano systems for biomedical applications, Springer, (2007) 67-81.
- I.F. Uchegbu, The biodistribution of novel 200 nm palmitoyl muramic acid vesicles, Int. J. Pharm. 162 (1998) 19-70.
- I.F. Uchegbu, A. T. Florence, Non-ionic surfactant vesicles (niosomes)-physical and pharmaceutical chemistry, Adv. Colloid Interface Sci. 58 (1995) 01–55.
- Rajnish Arora, C. P.Jain, Advances in Niosome as a Drug Carrier: A Review, Asian. J.Pharm. 1(2007) 29-39.
- EK Weibel, P Hadvàry, E Hochuli, E Kupfer, H Lengsfeld, Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. I. Producing organism, fermentation, isolation and biological activity, J Antibiot . 40 (1987) 1081-1085.
- Ballinger anne, R. Peikin Steven, Orlistat: its current status as an anti-obesity drug, Eur. J. Pharmacol. 440 (2002) 109– 117.
- E.Suzuki, High-resolution scanning electron microscopy of immuno gold-labelled cells by the use of thin plasma coating of osmium, Journal of Microscopy, 208 (2002) 153–157.
- Susan S. D Souza, P. Patrick DeLuca, Development of a Dialysis in- Vitro Release Method for Biodegradable Microspheres, AAPS PharmSciTech. 2005; 6(2): E323- E328.
- B. Shi, C. Fang, Y. Pei, Stealth , PEG-PHDCA niosomes: effects of chain length of PEG and particle size on niosomes surface properties, in vitro drug release, phagocytic uptake, in vivo pharmacokinetics and antitumor activity, J. Pharm. Sci. 95 (2006) 1873– 1887
- Luypaert J Zhang, M.H. Massart D.L, Feasibility study for the use of near infrared spectroscopy in the qualitative and quantitative analysis of green tea, Camellia sinensis (L.), Analytica Chimica Acta, 478 (2003) 303–312.
- Yoshioka T, Sternberg B, Florence AT. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (span 20, 40, 60, 80) and a sorbitan triester (span 85), Int J Pharma, 105 (1994) Y6.
- Michael E Aulton, Pharmaceutics the science of dosage form design. 2nd Ed 23(2002) 65- 73.
- RH. Muller, Ratrin Peter, Nanosuspension for the formulation of poorly soluble drugs- preparation by size reduction I-technique, Int. J. Pharm. 160 (1998) 229-237.
- H. Leiberman, Lachman, J. Schwartz, pharmaceutical dosage forms, Tablets 2, 2nded, marcel decer , newyork, 2 (1990), 229.
- MN. Azmin, AT. Flourence, RM. Handjani- Vila, FB. Stuart, G.Vanlerberghe, Whittaker JS, The effect of non-ionic surfactant vescicles (niosomes) entrapped on the absorption and distribution of methotextrate in mice, J. Pharm. Pharmacol. 37 (1985) 237-242.


