Allam Fayez Abuhamda1* and Aymen Elsous2
1Gaza Strip’s Neonatal Intensive Care Units, Ministry of Health, Gaza Strip, Palestine
2Department of Health Professionals, Israa University, Gaza Strip, Palestine
*Corresponding Author:
Allam Fayez Abuhamda
Senior Consultant Neonatologist
Gaza Strips Neonatal Intensive Care Units
Ministry of Health, Gaza strip, Palestine
Tel: +00972597502720
E-mail: allam570@yahoo.com
Received date: March 14, 2016; Accepted date: April 19, 2016; Published date: April 22, 2016
Citation: Abuhamda AF, Elsous A (2018) Oral-Facial-Digital Syndrome Type II (Mohr Syndrome) in Palestine. Ann Clin Lab Res Vol.6 No.4: 259. doi:10.21767/2386-5180.100259
Keywords
Oral-facial-digital syndrome; Polydactyly; Syndactyly; Cleft upper lip; Cleft hard palate
Introduction
Oral-Facial-Digital Syndrome (OFDS) is a congenital disorder characterizes with a variety of clinical abnormalities including: hypotelorism, hypertelorism, wide nose bridge, cleft lip, cleft palate, microglossia, micrognathia, retrognathia, preaxial polydactyly, postaxial polydactyly, clinodactyly, polycystic kidney disease and diverse forms of CNS defects resulting in different degrees of mental retardation, delay of speech and motor control deficit. There is at least 13 types of OFDS and each form is classified according to the existing anomalies. OFDS could be autosomal dominant, autosomal recessive, Xlinked dominant or X-linked recessive [1,2]. The OFD1 (Oral- Facial-Digital Syndrome Type I) gene provides instructions for making a protein whose function is not fully understood. It appears to play an important role in the early development of many parts of the body, including the brain, face, limbs, and kidneys. Mutations in the OFD1 gene prevent cells from making enough functional OFD1 protein, which disrupts the normal development of these structures [3,4]. Diagnosis of OFD syndrome type I when suspected, may be confirmed by genetic testing [5]. We present two cases of OFDS type II, which is a rare autosomal recessive disorder [6] giving that the gene responsible is still unknown and the incidence is 1 per 3 lakhs live births [7,8].
Case Reports
Case 1
The parents are cousins and mother was G3P1. The first pregnancy at 30 weeks of gestational age as a result of intrauterine fetal death. The fetus was female and presented with dysmorphic features. She had polydactyly, syndactyly, cleft palate and cleft lip. Same thing happened to the second pregnancy and typically at same gestational age, however, the female baby showed normal physical examination.
A full term first sibling baby of family, a female baby born through normal vaginal delivery and her birth weight was 2800 mg. Her Apgar score was 7 and 9 at 1 and 5 minutes respectively. Findings from physical examination after birth were: small eyes, broad nose bridge, midline cleft upper lip, microglossia, cleft palate and micrognethia. Both hands have clinodactyly and preaxial polydactyly as there are 7 fingers (duplicate thumb). There is also syndactyly between thumbs (bifid thumb) in each hand. Both feet have Talipes equinovarus and preaxial polydactyly as there are 7 toes (duplicate big toe). Furthermore, a syndactyly between two big toes (Bifid big toes) (Figures 1-6). She had normal external female genitalia. She was hypotonic but able to move all limbs symmetrically. The respiratory system function and hemodynamic status were wise stable (Figure 7). Brain ultrasound, abdominal ultrasound, and echocardiography were all normal. One day after birth, we started feeding on orogastric tube, and she tolerated the feedings and passed stool normally (Figure 8). She was discharged home at the age of 3 days in a good general condition and referral was written for consultations to maxillofacial, hands, orthopaedic and plastic surgery.
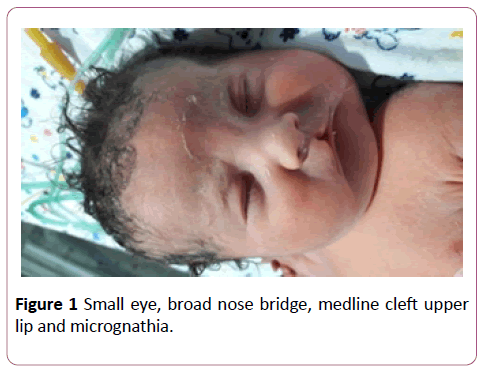
Figure 1: Small eye, broad nose bridge, medline cleft upper lip and micrognathia.
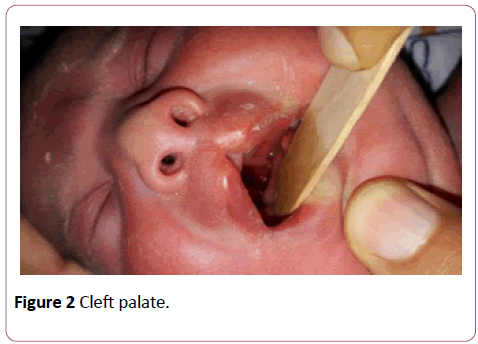
Figure 2: Cleft palate.
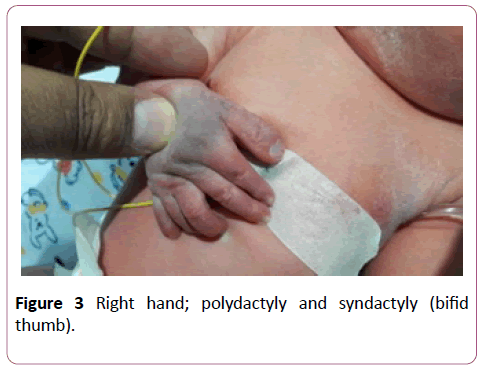
Figure 3: Right hand; polydactyly and syndactyly (bifid thumb).

Figure 4: Left hand; polydactyly and syndactyly (bifid thumb).
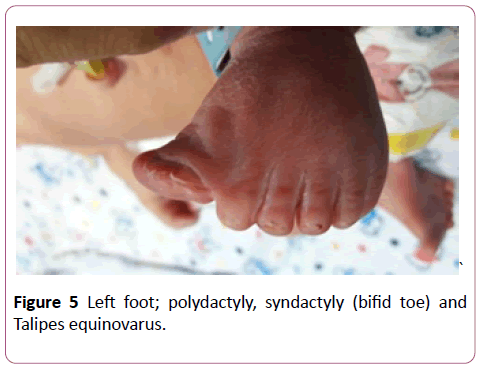
Figure 5: Left foot; polydactyly, syndactyly (bifid toe) and Talipes equinovarus.
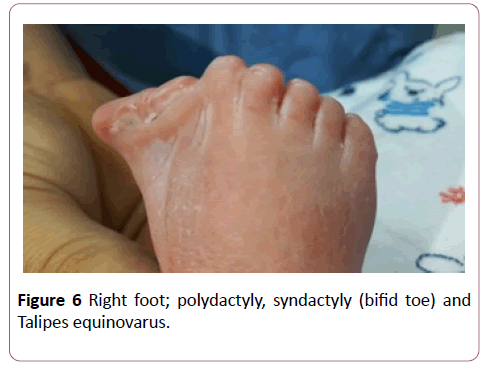
Figure 6: Right foot; polydactyly, syndactyly (bifid toe) and Talipes equinovarus.

Figure 7: Congenital hypotonia.
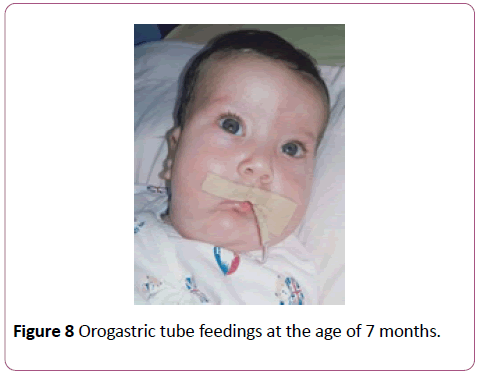
Figure 8: Orogastric tube feedings at the age of 7 months.
Case 2
Parents were cousins and had not previous family history of any congenital anomalies. The male baby was their first sibling and had been first seen at the age of 4 years. By physical examination, the child had some abnormal features; Antimongoloid eye slant, prominent ears, broad nose bridge (Figure 9), high arched palate, cleft soft palate, lingual nodule (Figure 10), preaxial and postaxial polydactyly with syndactyly in both hands, bilateral bifid thumbs and bilateral clinodactyly (Figure 11). Both foot had polydactyly with syndactyly, and bifid big toe shape (Figures 12 and 13). The child had a clear micropenis and undescended testes (Figure 14). Karyotyping study result showed normal study (46, xy). Brain CT, echocardiography and abdominal renal ultrasonography were also normal. The baby suffered from a global developmental delay. Similar dysmorphic features appeared to the second sibling male baby, however he passed away at age of 2 months.

Figure 9: Anti-mongoloid eye slant, prominent ears, and broad nose.
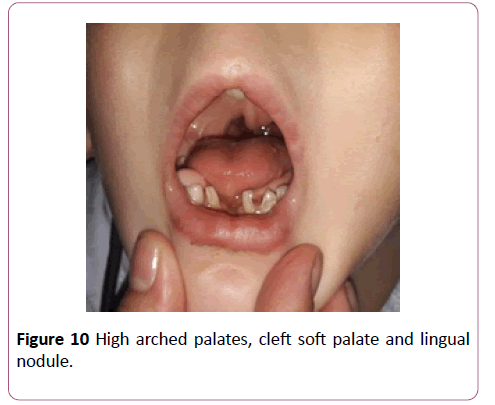
Figure 10: Anti-mongoloid eye slant, prominent ears, and broad nose.
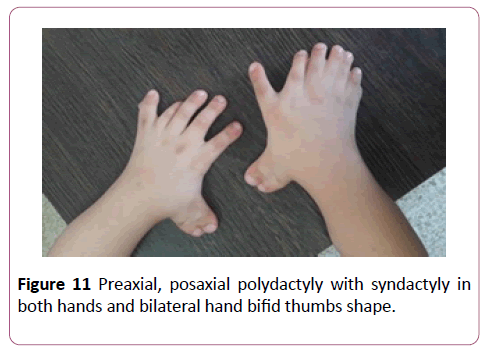
Figure 11: Preaxial, posaxial polydactyly with syndactyly in both hands and bilateral hand bifid thumbs shape.

Figure 12: Left feet; feet had preaxial, posaxial polydactyly with syndactyly and bifid big toe shape.
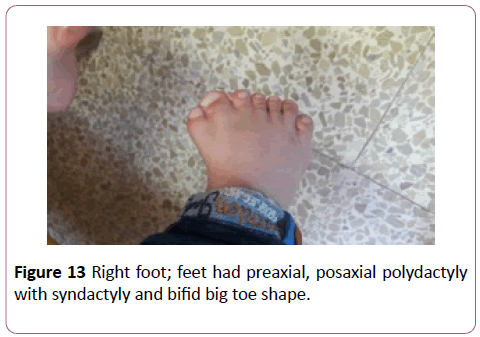
Figure 13: Right foot; feet had preaxial, posaxial polydactyly with syndactyly and bifid big toe shape.

Figure 14: Micropenis and undescended testes.
Discussion
In both cases, the parents were consanguineous [9] and had characteristic features of oral facial digital syndrome type II which include; broad nose bridge, high arched palate, cleft palate, lingual nodule, hands had preaxial, postaxial polydactyly with syndactyly in both hands, and bifid thumb shape. Also feet preaxial, postaxial polydactyly with syndactyly and bifid big toe shape. Oral-facial-digital syndrome (OFDS) type II (Mohr Syndrome) can be diagnosed from the medical history and physical examination of the patient [10-12]. Both cases required consultation and follow up with plastic surgery, maxillofacial surgery, hand surgery for reconstruction and repair of the anomalies. Moreover, follow up with audiology and dentist was applied. The 2nd case also needed follow up with pediatric surgery regarding undescended testes. Both cases need; multidisciplinary team approach to follow up different complaints, to coordinate between different medical specialties, and to observe the global development [13].
Conclusions and Recommendations
• Two rare cases were presented with symptoms of oral facial digital syndrome type II in Palestinian children.
• Consanguineous marriage in Palestinian people carries a high risk for congenital anomalies. Therefore, it is recommended to avoid relative marriage as possible.
Ethical Approval
Hospital permission was obtained to review infant's medical file and collect the necessary information. Parental/guardian consent obtained to set for interview and provide additional information not found in infant's medical file, and to show baby's whole face.
Availability of Data
Data is available and under request with first author
Conflict of Interest
The authors declare no conflict of interests.
23502
References
- Fiorella G, Franco B, Toriello H, Neri G (2007) Oral digital syndromes: A review and diagnostic guidelines. Am J Med Genet 143 (24): 3314-3323.
- Anneren G, Gustavson KH, Jozwiak S, Kjartansson S, Str ƒmberg B (1990) Abnormalities of the cerebellum in oro-facio-digital syndrome II (Mohr syndrome). Clin Genet 38(1): 69-73.
- Brunella F, Thauvin-Robinet C (2016) Update on oral-facial-digital syndromes (OFDS). Cilia 5(1): 12.
- Lopes CA, Prosser SL, Romio L, Hirst RA, O'Callaghan C, et al. (2011) Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J Cell Sci 124(4): 600-612.
- Toriello HV, Franco B, Bruel AL, Thauvin-Robinet C (2016) Oral-facial-digital syndrome type I. Gene Reviews [Internet].
- Alessandro SC, Dallapiccola B (2005) Oro-facial-digital Syndrome, Type II. Abnormal skeletal phenotypes: From simple signs to complex diagnoses pp: 787-790.
- Biswas A, Ghosh JK, Sinha MK, Basu K, Chatterjee S (2009) Mohr-Claussen syndrome or oro-facial-digital syndrome (OFDS) type-II. J Pak Med Assoc 59(7): 484-486.
- Ahmad ST (2010) Genetic disorders among Arab populations. Springer Science & Business Media.
- Ajacques JC (1981) Mohr's syndrome: Type II orofaciodigital syndrome (author's transl). Revue de stomatologie et de chirurgie maxillo-faciale 82(4): 234-240.
- Sakai N, Nakakita N, Yamazaki Y, Ui K, Uchinuma E (2002) Oral-facial-digital syndrome type II (Mohr syndrome): Clinical and genetic manifestations. J Craniofac Surg 13(2): 321-326.
- Ouwens M, Wollersheim H, Hermens R, Hulscher M, Grol R (2005) Integrated care programmes for chronically ill patients: A review of systematic reviews. Int J Qual Health Care 17(2): 141-146.