Keywords
|
| RB, Tumor suppressor, E2Fs, Cell cycle, Differentiation, Metabolism |
Introduction
|
| The loss-of-function mutation in the RB tumor suppressor gene at tumor initiation occurs in surprisingly few types of cancer. The inactivation of the RB product is often found during the progression of common types of cancers including prostate, breast, bladder, and esophageal cancers [1]. Most canonical function of pRB is to control cell proliferation that is achieved by preventing inappropriate entry of the cell into the cell cycle. This was experimentally confirmed by cell cycle re-entry upon RB inactivation in several tissues [2-4]. |
| In vivo analyses of genetically engineered mice and in vitro experiments contributed to determining many of the canonical functions of pRB in cell cycle control and differentiation. During embryonic hematopoiesis, the loss of RB in mouse embryos results in inefficient enucleation and incomplete terminal differentiation of erythroid cells [5,6]. During skeletal muscle development, pRB is required for the cells to properly exit the cell cycle and to complete differentiation [7]. A myogenic transcription factor, MyoD, activates expression of pRB and Cdkn1a (p21) to enforce cells to exit the cell cycle [8]. pRB is also required for reorganization of the lamin speckles during the myogenic process [9]. In the intestine of mice, when RB is conditionally deleted, differentiation markers showed abnormal patterns, and proliferative crypt cells exhibited enhanced proliferation [10,11]. In the lens, the loss of RB altered the expression of genes promoting differentiation, including β- and γ-crystallines [12]. These deficiencies in differentiation following pRB inactivation seem at least partially due to a defect in exiting the cell cycle, which is a critical step for most differentiation processes [1]. In addition, pRB controls the pluripotency of cells, independent of the cell cycle [13,14]. Therefore, pRB is not only a cell cycle regulator, but also a key factor that controls cellular dedifferentiation and transformation. pRB is also implicated in numerous varieties of biological events such as cell death, DNA damage response, cellular senescence, genomic instability, cellular metabolism, inflammation, and angiogenesis [15-19]. |
| In this review, in as much as space allows, we describe the molecular bases for the pleiotropic functions of pRB and its family members p107 and p130, which involve upstream signaling, effectors, and context-dependent targeted genes. |
Regulation of G1-S Transition by pRB
|
| pRB has domain structures named pocket A, B, and C [20]. These domains individually or in combination contribute to the physical binding of many factors (effectors) involved in cell proliferation, transcriptional regulation, chromatin modification, signal transduction, apoptosis, etc. Therefore, canonically, pRB has been called a pocket protein. On the other hand, the reason for this gene being called an adaptor protein is that pRB undergoes various post-translational modifications by different types of signals and this process alters its binding affinity to effectors [17]. |
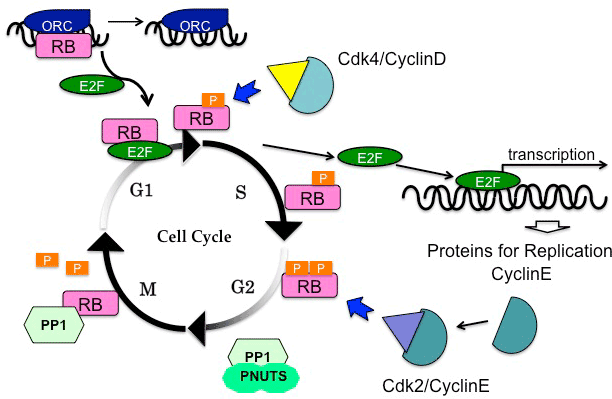
| The most common post-translational modification on pRB that alters its functions is phosphorylation by kinase complexes consisting of CDK4 or CDK6 and cyclin D, which occurs in late G1 phase to early S phase (G1/S transition), and the subsequent release of E2F transcription factors promotes progression to the S phase [21]. The E2F family proteins are classified into activators including E2F1, E2F2, and E2F3, and into repressors including E2F4 and E2F5. E2F6, E2F7, and E2F8 have been additionally identified, and currently being analyzed. It is also possible that E2F4 and E2F5 activate gene transcription under certain conditions [22]. Especially, E2F4 may transactivate several genes whose products are involved in the G2/M phase [23-25]. During the G1 phase, pRB forms a transcriptional repressive complex with histone deacetylase (HDAC) and the nucleosome remodeling complex hSWI/SNF. This machinery primarily represses the expression of cyclin E and A genes. Phosphorylation of pRB by CDK4/cyclin D allows HDAC to dissociate from the complex, relieving repression of cyclin E and thereby promoting cell cycle entry into the S phase. However, the phosphorylated pRB and hSWI/SNF complex persists on the promoter of genes encoding cyclin A and CDC2, inhibiting cell cycle exit from the S phase [26]. These processes explain the sequential expression of cyclin E and A during the cell cycle. |
| There are 13 amino acid residues in the pRB protein that are possibly phosphorylated by CDK4 or CDK6 [27]. In these phosphorylation sites, the 608th serine residue (S608) that resides between pocket A and B determines the binding affinity to the transactivation domain of E2F1, E2F2, and E2F3. These factors transactivate genes implicated in cell cycle progression and nucleotide synthesis such as CDC6, cyclin E, replication protein C, DNA ligase, and DNA topoisomerase [23,28-30]. Cell cycle and bone development defects in RB-deficient mouse embryos were suppressed by the simultaneous deletion of E2F1 [31]. In addition, numerous reports have indicated that E2Fs play a key role in mediating pRB function to regulate cell cycle and differentiation. |
| Besides the E2F-mediated transcriptional control, pRB has alternative mediators to regulate DNA replication. Hypophosphorylated pRB specifically binds to the largest subunit of the origin recognition complex (Orc1); this interaction competes with E2F1 on binding to pRB (Figure 1). During the transition from the G1 phase to the S phase, E2F1 out-competes Orc1 from pRB at replication origins [32]. The functional relationship with Orc1 represents one of the E2F-independent functions of pRB. |
| RB+/- mice developed pituitary and thyroid tumors as a consequence of biallelic loss of RB in somites. Simultaneous deletion of E2F1 significantly suppressed thyroid tumorigenesis [33]. In contrast, deletion of E2F3 accelerated malignant progression of RB-deficient thyroid tumor [34]. Interestingly, RBD326V/+ mice develop pituitary but not thyroid tumors. This observation is in line with the result of an in vitro binding assay indicating that mutated pRBD326V could bind to E2F1 but not to E2F2/3 [35]. Thus, the relationships between pRB and each of E2F family members are functionally distinct. |
| On the other hand, the introduction of the RB D750F/D750F mutation, which is able to inhibit E2F transactivation activity but unable to interact with LxCxE-motif proteins, including HDACs, heterochromatin protein 1 (HP1), histone methyltransferase Suv39h1, and CtBP-interacting protein, caused cell cycle arrest upon mitogen deprivation or cell-cell contact, but did not cause cell cycle arrest upon RasV12 introduction or irradiation [36]. This result supports the argument that pRB-binding partners determine cell behaviors that differ depending on cellular context. |
Regulation of Mitosis by pRB
|
| In addition to phosphorylation, dephosphorylation of pRB is crucial for cell cycle control, especially during mitosis (Figure 1) [37]. Upon exiting mitosis, pRB is dephosphorylated by protein phosphatase 1 (PP1) [38]. In fact, direct binding between PP1 catalytic subunit (PP1c) and pRB has been observed during mitosis [39]. Before initiating pRB phosphorylation, CDK/cyclin complexes out-competes PP1 [40]. Moreover, PP1 nuclear targeting subunit (PNUTS) specifically inhibits the activity of PP1 to dephosphorylate pRB [41,42]. |
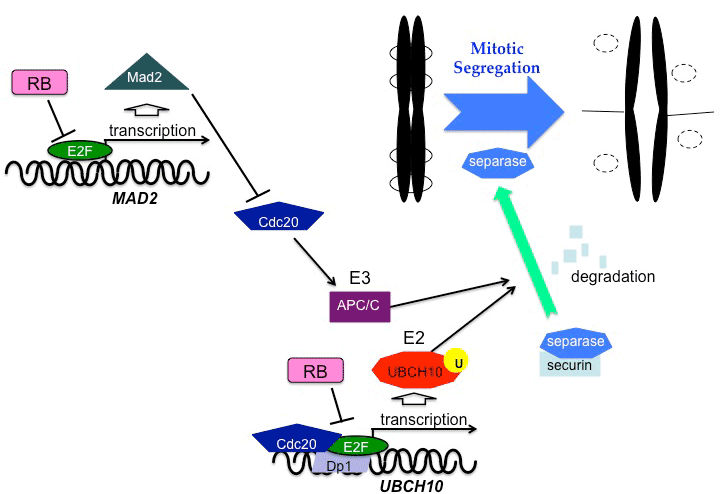
| pRB is involved in checkpoints and maintenance of mitosis as well as in the transition from the G1 phase to the S phase. Chromosomal instability accompanied by abnormal spindle formation and impaired cohesion were also observed in pRB-deficient cells [43,44]. The expression of Mad2, a component of the mitotic checkpoint, is induced by E2Fs released from pRB [45]. Mad2 ensures proper progression to anaphase, as it blocks anaphasepromoting complex/cyclosome (APC/C). An aberrant expression of Mad2 caused by pRB defects may generate a hyperactive response to spindle checkpoint, causing an abnormal order of mitotic events and low accuracy of chromosomal segregation. Persistent inactivation of the APC/C by excess amounts of Mad2 may delay the degradation of securin and cyclin B. Cdc20 protein, which is an activator of APC/C, functions as a co-transactivator of the UBCH10 gene, which encodes the E2 ubiquitin carrier protein [46]. Cdc20 interacts with the E2F1-DP1 complex at the promoter region of UBCH10 to stimulate its transcription, and pRB represses this transactivation [47]. pRB inactivation leads to deregulation of UBCH10 expression, which may cause lower accuracy of chromosomal segregation (Figure 2). Moreover, pRB directly binds to the nuclear mitotic protein, NuMa, which organizes the mitotic spindle [48]. Since this binding occurs during the transition from interphase to prophase, it may control timing of the spindle formation or prevent mitotic segregation caused by premature chromosomal arrangement. |
| Moreover, pRB controls genomic stability. Malignant tumor progression is caused by pRB-deficiency, which induces aneuploidy and genomic instability [49]. It is also observed in cultured cell lines upon pRB inactivation [45,50-52]. Furthermore, hepatitis C virus (HCV) infection, which often causes polyploidy, decreased the effects of mitotic checkpoints due to the reduction of RB transcription and the induction of E2F1 and Mad2 [53]. Loss of p53 seems to render the cells resistant to cell cycle arrest, following chromosomal mis-segregation induced by pRB deficiency. Therefore, simultaneous inactivation of pRB and p53 results in high levels of chromosomal instability [54]. The functional synergy between pRB and p53 will be discussed again later. |
Post-translational Modification of pRB
|
| The pRB-E2F1 complex plays a pivotal role in regulating apoptosis, in a manner distinct from that for transcriptional control during the cell cycle [55-58], and for enhancing resistance to phosphorylation by CDK [59,60]. Specific phosphorylation of human E2F1 at S364 that occurs in response to DNA damageinduced double strand breakages enhances the formation of the pRB-E2F1 complex, even when pRB is hyperphosphorylated; this process subsequently transactivates a number of proapoptotic genes [55]. |
| Acetylation of pRB by p300 is required for cell cycle exit during skeletal myogenesis in mice [61]. In addition, acetylation of pRB at K873 and K874 in the C-terminus region of pRB, where E2F1 binds to, occurs in response to DNA damage, which releases E2F1 from pRB [62]. |
| Furthermore, methylation at K810 in pRB by the Set7/9 methyltransferase, which prevents subsequent phosphorylation of pRB, is required for efficient cell cycle arrest [63]. This methylation inhibits the phosphorylation of serine residues S807 and S811 in the vicinity. Phosphorylation at these serine residues by 5’-AMPactivated protein kinase (AMPK) is required for proliferation of neural stem and progenitor cells (NPC) in the mouse brain [64]. In some lineage of cells including NPC, metabolic stimuli to control cell proliferation are mediated through the AMPK-pRB pathway rather than through the CDK-pRB pathway. On the other hand, a putative oncoprotein, SET and MYND domain-containing protein 2 (SMYD2), whose overexpression is often observed in esophageal squamous cell carcinoma cells, also methylates K810 in pRB, and the methylation by SMYD2 facilitates phosphorylation at S807 and S811 by CDK4 [65]. The growth suppression of cancer cells by SMYD2 knock-down is possibly due to the altered methylation in pRB. |
| In endothelial cells exposed to oxidative stress, rapid dephosphorylation of the pRB family of proteins was observed, which appeared to depend on protein phosphatase 2A (PP2A); this enhances pRB-E2Fs association [66]. UV stress caused the rapid dephosphorylation of p107 by PP2A holoenzymes [67,68]. However, growth arrest by FGF or all-trans retinoic acid was mediated by dephosphorylation of p107 or p130 in a PP2A-dependent manner [69-71]. Dephosphorylation at two residues, S1080 and T1097, adjacent to the nuclear localization signal (NLS) in p130 induced its translocation to the nucleus due to increased binding affinity of importins to this NLS [72]. Moreover, hypoxia (1% oxygen) and oxidative stress caused dephosphorylation of the pRB family proteins by PP1 and PP2A, which reduces the efficiency of DNA replication [66]. Thus, the phosphorylation status of pRB and its family members are dynamically controlled by the equilibrium between CDKs and PP1 or PP2A throughout the cell cycle. A recent study using chemotherapeutic reagents suggested that PP1 but not PP2A is directly involved in dephosphorylation of pRB. PP2A might affect the phosphorylation status of PP1c and/or its regulatory proteins [73]. These findings suggest that the effects of post-translational modifications in pRB and other pocket proteins differ depending on cellular context. |
Epigenetic Regulation by pRB
|
| As aforementioned, pRB controls its targeted gene transcription mainly via E2Fs. Genes targeted by E2Fs include those promote S phase, chromatin assembly, condensation, segregation, and spindle formation [25], but a considerable percentage of those genes might be regulated through pRB function to modulate the epigenetic status of the genome [74]. The pRB-E2F complex suppresses gene transcription at least partly by recruiting HDAC1 and 2 [75]. Histone H4 acetylation status on E2F targeted promoters varies in a cell cycle-dependent manner [75-77]. pRB interacts with the histone methyltransferases and HP1 [78,79]. Loss of pRB resulted in disappearance of trimethylated K20 histone H4 from the heterochromatin domain [80]. As described above, pRB interacts with HDAC and the hSWI/SNF complex to repress expression of cyclin E and A during the G1 phase. |
| In addition, pRB controls the methylation status of the genome, by binding to DNA methyltranferase 1 (DNMT1). Through this interaction, pRB reduces DNA methylation levels in the genome [81], while DNMT1 cooperates with pRB-E2Fs to suppress transcription via HDAC1 [82]. |
| There are several alternative pathways where pRB controls DNMT1 activity. Attenuation of pRB expression stimulates ATM expression depending on E2F, whereas pRB appeared to cooperate with ATM to regulate the stability of DNMT1 by controlling its complex formation with a specific E3 ligase UHRF1 [83]. On the other hand, through direct binding at the AP-1 site, pRB and c-JUN upregulate the DNMT1 promoter in early passage mesenchymal stromal cells (MSCs) [84,85]. In non-MSCs, pRB was found to bind to the E2F1 binding site but not to the AP-1 site in the DNMT1 promoter [84], implicating that the functional interactions between pRB and DNMT1 might be critical for epigenetic regulation that determines lineage commitment and the differentiation status of cells. |
pRB Functions To Control Cellular Metabolism
|
| pRB is deeply implicated in the regulation of cellular metabolism [18]. This would be highly helpful to adjust the metabolic status to demands to increase biomass upon cell proliferation. Cancer cells are most remarkably characterized by altered cellular metabolism. Most cancer cells achieve aerobic glycolysis that is inefficient to generate ATP, while normally differentiated cells rely much on oxidative phosphorylation to generate energy. This phenomenon is called the Warburg’s effect. |
| The role of pRB in cancer metabolism is being extensively studied recently [18,86]. The pRB-E2F pathway regulates cellular proliferation in response to glucose stimulation. E2F1 induces the expression of Kir6.2, which constitutes the ATP sensitive potassium channel, thereby regulates insulin release from pancreatic-cells. Meanwhile, CDK4 is activated by the insulin signal in response to glucose stimulation, resulting in activation of E2F1 [87]. |
| pRB is implicated in controlling many of the genes involved in fatty acid and cholesterol biosynthesis. The promoters of these genes possess sterol regulatory elements (SRE), E2F-binding consensus sequences, or both. RB inactivation causes an E2Fdependent induction of farnesyl diphosphate synthase, several prenyltransferases, and transcriptional activator sterol regulatory element-binding proteins (SREBPs), thus the isoprenylation status of proteins including N-Ras is affected by the presence of pRB [88]. |
| While human cancers without functional pRB exhibits an increased glutamine-uptake, triple knock-out of the RB family of genes in mouse embryonic fibroblasts (MEFs) also increased glutamine consumption, due to upregulation of the glutamine transporter ASCT2 [89]. Loss of RB family members resulted in higher glutamine utilization in the TCA cycle and glutathione accumulation. Moreover, these cells became more dependent on glutaminolysis [89]. Thus, pRB family members may play a key role in rewiring glutamine metabolism and glutathione synthesis in tumor cells. |
| pRB and E2F1 can also regulate oxidative metabolism by modulating the expression of several genes involved in mitochondrial biogenesis [90,91]. During differentiation of white and brown adipocytes that takes place depending on the status of pRB, mitochondrial gene expression pattern and number are altered. The pRB-E2F1 complex binds to the promoters of many of the genes implicated in oxidative metabolism in brown adipose tissue and muscle. When exposed to cold or fasting conditions, pRB undergoes higher rates of phosphorylation, stimulating oxidative metabolism [90,92]. Thus, pRB detects changes in the extracellular space and directs cell metabolism in response. |
pRB in tumor progression
|
| Mice carrying a point mutation in RB that impedes the ability to interact with the transactivation domain of E2Fs showed relatively normal development and did not develop cancer throughout their lives [60]. Interestingly, MEFs with a pRB mutation that specifically disrupts interactions with E2Fs, were able to arrest the cell cycle upon serum starvation at an efficiency similar to that of wild type MEFs, but their capability of progressing the cell cycle upon serum stimulation was impaired to a level similar to that of RB knock-out cells [60]. In short, cell cycle arrest can be achieved by pRB without the help of E2Fs. These results suggest that the pRB function in transcriptional control via the interaction with E2Fs is distinguished from the pRB function in tumor suppression. Therefore, pRB deficiency gives rise to a variety of consequences due to its pleiotropic functions that differ depending on cellular types and stages of tumor development. For example, heterozygosity of retinoblastoma gene RB+/- tends to arise thyroid C cell tumors, but the RB+/- Skp2-/- mice did not appear the tumor in thyroid glands. Elimination of p27 phosphorylation, which avoid ubiquitination by SCFskp2, showed the same effect given by the Skp2-null background [93]. This suggests that downregulation of p27 by ubiquitination under the SCFSKP2 system supported aberrant proliferation in RB-deficient cells. |
| RB mutations in early stage tumorigenesis are detected in certain types of malignancies such as retinoblastoma, osteosarcoma, and small cell lung cancers. In the majority of cancers, pRB inactivation is more prevalent during the later stage of tumor development. pRB is uniquely indispensable to prevent precursor proliferation of cones in the retina, implicating absolute requirement of RB dysfunction for the initiation of retinoblastoma [94]. On the other hand, pRB is typically intact at early stages of prostate carcinogenesis. The inactivation of pRB at later stages allows E2F1 to activate the expression of androgen receptor, which is essential for tumor progression and metastasis [95]. Furthermore, the pRB downregulation reduces the expression level of E-cadherin; this gives rise to a mesenchymal-like phenotype in breast cancer cells thereby promotes invasion and metastasis [96]. |
Functional Synergy between pRB and p53
|
| pRB and p53 are the targets for the proteins produced by oncogenic DNA viruses. For example, the E7 protein of human papillomaviruses (HPVs) binds to pocket proteins, pRB, p107, and p130, synergistically inhibiting their functions [97]. HPV E7 also binds to and activates E2F1; this blocks E2F6 function to counteract E2F1 function [98]. On the other hand, the E6 protein of HPVs stimulates ubiquitination of p53 [99]. These findings indicate that the dual inhibition of pRB and p53 by HPV proteins promotes cancer initiation. |
| Apoptosis and cellular senescence are crucial processes to avoid malignant progression, and are mostly controlled by p53, pRB in cooperation with other tumor suppressors. p53 and pRB cooperate in multiple steps in these mechanisms, suggesting why double mutation of these genes is often detected in cancers. Loss of pRB function can lead to apoptosis mediated by E2F1 and then by ARF, an upstream regulator of p53 [100,101]. Thus, pRB functions in a considerably different way in the absence of p53. Knockdown of either one of p53, p21, or pRB restored proliferative properties in cells, which were arrested by loss of the three ras loci, H-, N- and K-ras, implicating pRB-p53-p21 function in the identical process [102,103]. However, a recent study demonstrated that a double inactivation of pRB and p53 resulted in an undifferentiated status in MEFs without inducing carcinogenesis [104]. This phenomenon was not fully achieved in cells deficient of either one of pRB or p53, indicating that their functions acted in distinct pathways |
| A considerable portion of pRB proteins localize to the mitochondria and play pivotal roles in regulating mitochondrial apoptosis in a transcription-independent manner [105,106]. Surprisingly, this regulation is attained via the direct interaction of pRB with one of the Bcl2 protein family members, Bax [106]. Bax increases permeability of the mitochondrial outer membrane (MOMP), releasing proapoptotic factors such as cytochrome c and leading to activation of effector caspases [107]. The status of mitochondrial pRB was affected by a wide variety of proapoptotic signals, which is one of the non-canonical functions of pRB in tumor suppression [106]. Phosphorylation of pRB at S807 appeared to be critical for the interaction with Bax [108]. Hence, dephosphorylation of pRB results in its dissociation from Bax. However, knockdown of PNUTS, which would activate PP1 that dephosporylates S807 in pRB, induced apoptosis [109-111]. It is thus unclear whether release of Bax from pRB is required for mitochondrial apoptosis. On the other hand, dephosphorylation of pRB at T821 that occurs in response to cellular stress, such as UV or CDK inhibitors, stimulated apoptosis [112,113]. Whether the phosphorylation status of pRB at T821 affects the binding between pRB and Bax is not yet clarified. |
| In mitochondrion, p53 also directly interacts with anti-apoptotic proteins such as Bcl2 or Bcl-XL. This interaction results in activation of proapoptotic Bax and Bak proteins [114]. The overlapped pRB and p53 functions in controlling apoptosis would explain the necessity for inactivation of pathways related to both tumor suppressors for tumorigenicity [115,116]. Interestingly, the non-cell autonomous function that suppresses apoptosis in RB deficient cells occurred after p53 induction in neurons of chimeric mice [117]. It may caused by some survival signals or Bax inhibition from neighbor cells. Moreover, a study using tetraploidy technique showed an extraembyonic function of the RB gene. RB-/- embryos, when supplied with wild-type placenta, did not show abnormal development in the central nervous system (CNS), and survived until birth, however died after birth due to severe skeletal muscle defects [118,119]. In lens development, RB, however, appeared to function in a perfectly cell autonomous manner [119]. Therefore, during embryonic development, pRb could function in both non-cell and cell autonomous manners. Molecular aspects of the non-cell autonomous phenomena in RB-/- cells during development are remained to be elucidated. |
|
Pocket Proteins under the Control of Transforming Growth Factor β
|
| Transforming growth factor (TGF) β exerts tumor-suppressing activities at least partially via the regulation of MCM7. The helicase activity of this molecule is directly inhibited by pRB [120-122]. The MCM complex is recruited to origin recognition complexes to initiate transcription through its helicase activity in early S phase. The MCM7-pRB interaction mediated by TGFβ prevents pre-replication complex activation by the MCM complex until the G1/S boundary is reached [122]. TGFβ stimulates E2F4/5 to form complexes with HDAC and pocket proteins p107 or p130, resulting in the expression of c-MYC or CDC25A, respectively [123,124]. Overexpression of MCM7 is observed in a wide range of cancers. Furthermore, suppression of MCM7 decreased BrdU incorporation in lung and bladder cancer cells [125]. Additionally, TGFβ stimulates E2F1 to form a complex with p300/CREBbinding protein-associated factor (P/CAF), allowing pRB to induce proapoptotic proteins [126]. The E2F1-pRB-P/CAF complex also induces proapoptotic proteins such as p73 and caspase7 in response to DNA damage [57]. Thus, pRB in cooperation with its family members plays a pivotal role in deciding whether to allow cells to go through apoptosis or to progress through the cell cycle. |
pRB in Cellular Senescence
|
| The formation of senescence-associated heterochromatin foci (SAHF) contributes to cell cycle exit by directly silencing proliferation-promoting genes [127-130]. During the SAHF formation, pRB cooperates with histone chaperones HIRA/ ASF1a [131]. Phosphorylation of HIRA and HP1 promotes SAHF formation by recruiting macroH2A, HP1γ, or HMGA [130,131]. pRB and p16INK4a are indispensable for SAHF formation induced by oncogene Ras, but p53 and p21CIP1 are dispensable [128,132]. In addition, the induction of SAHF in response to DNA-damage required the mitogen-activated protein kinase p38 [133]. It is also notable that pRB is required for the senescence-associated loss of trimethylated K4 histone H3, which is dependent on H3K4 demethylases including Jarid1a and Jarid1b [134]. |
| Activation of oncogene products such as Ras triggers cellular senescence, a state of irreversible cell growth arrest when cells are mostly intact; this is called oncogene-induced senescence (OIS) [127,135]. In OIS, pRB and E2Fs colocalize to Promylocytic leukemia (PML) bodies and suppress E2F targeted promoters [136,137]. Of the E2F family members, only E2F7 is upregulated in OIS. Once induced, E2F7 represses expression of the E2F-targeted genes. Of the E2F family members, only E2F7 is capable of compensating for the loss of pRB to suppress mitotic genes [138]. |
| Knockdown of pRB in cells reversed the changes in gene expression during senescence and allowed them to reenter the cell cycle, while knockdown of p107 or p130 could not reverse the changes of senescence [139]. This effect represents an unique function of pRB in inducing cellular senescence. Yet, senescence induced by pRB inactivation depends on the function of p130 [88], suggesting redundant functions shared by pRB family members in controlling cellular senescence. |
Conclusion
|
| In this review article, we collected the published findings of the pleiotropic aspects of pRB function, and tried to connect them to the interaction with numerous binding partners. Because of the space limitation, we did not refer to the recent progress in our understanding of pRB functions in stem cells, inflammation, and other unexpected aspects of metabolic control. We propose that, in light of its strong clinical relevance, pRB should be continuously studied to elucidate its functions relating to different steps during carcinogenesis, which would be a valuable resource for cancer therapeutics. |
Acknowledgment and Grant Support
|
| We thank Dr. Shunsuke Kitajima for critical reading of this manuscript. This work was supported by Funding Program for Next Generation World-Leading Researchers (NEXT), Grantin- Aid for Scientific Research (MEXT), Astellas Foundation for Research on Metabolic Disorders, Takeda Science Foundation, Naito Foundation, Daiichi-Sankyo Foundation for Life Science, NOVARTIS Foundation (Japan) for Promotion of Science and Hokkoku Foundation for Cancer Research to CT. |
Figures at a glance
|
 |
 |
| Figure 1 |
Figure 2 |
|
| |
| |