Keywords
Polydatin; Phospholipid complex; Preparation; In vitro release; Pharmacokinetics
Introduction
As a traditional Chinese herbal medicine in Asia, polygonum cuspidatum root exhibits antibacterial, antioxidant and antiangiogenesis activities [1]. There are numerous bioactive components existed in polygonum cuspidatum root, such as emodin, resveratrol, polydatin, etc. [2-6]. Polydatin (3,4’,5-trihydroxystilbene-3-β-mono-D-glucoside, PD) (Figure 1), has a number of biological functions, including antiplatelet aggregation, anti-tumor activity, cardio-vascular diseases prevention and anti-oxidative activities [7-10]. Despite the promising pharmacological activities of PD, its poor aqueous solubility and low oral bioavailability have severely restricted its use in the clinic applications. Therefore, much effort has been done to overcome these shortages. A recent study showed that polydatin (PLD)-loaded liposome system was applied to promote the oral bioavailability and solubility of polydatin via the bioadhesive nature and a decreased particle size [11], although some limitations of PLD-loaded liposome system were still exposed, such as lower drug loading and poor stability. Recently, polydatin phospholipid complex has been developed and proved to be a novel and effective drug-carrier system with improved oral bioavailability.
Figure 1: The chemical structure of polydatin.
Soy phospholipid mainly consist of phosphatidylcholine, phosphatidylethanolamine and phosphatidylinositol, which are included in cell membrane with nontoxic, amphiphilic and endogenous properties [12]. Phospholipids play a major role in drug delivery due to their amphiphilic nature that can modify the solubility behavior and rate of drug release for the enhancement of drug absorption [13]. Most critically, phospholipids constitute the bilayer which is the basic skeleton of biomembrane, maintaining the cell membrane fluidity. On the basis of these biological properties of phospholipids, it is expected to make drug easier to penetrate cell membrane without disturbing the cellular lipid bilayer, further increasing the absorption and bioavailability of poorly soluble drug. In recent years, phospholipids have been widely used in drug delivery systems including niosomes, liposome, and solid lipid nanoparticles to enhance drug activity and reduce drug toxicity [14,15] or as emulsifiers in emulsion formulations [16].
Phospholipid complex (PC), as a potential technology of pharmaceutical preparations, was formed by intermolecular forces and electrostatic attraction of drug and phospholipids. In recent years, there is an upward trend in the study of phospholipid complex due to its positive impact in the improving of the solubility, stability, oral bioavailability and the therapeutic efficacy of poorly soluble drugs. several phospholipid complex of curcumin [17], Quercitin [18], kaempferol [19], Luteolin [20] and others, have been successfully prepared and improved solubility, bioavailability [21-23]. Maiti et al. reported that the AUC of curcumin-phospholipid complex was improved 7.57-fold after oral administration compared with curcumin [24].
The objective of this present study was to prepare PPC by solvent evaporation to improve the dissolution and bioavailability of PD. The physicochemical characterization of PPC was systematically investigated by FTIR, DSC, and XRD. Beside in vitro release experiments, oral administration in rats was employed to investigate in vivo oral bioavailability of PPC [25]. To our knowledge, there has been no report yet regarding to improving the bioavailability of PD by developing PPC.
Materials and Methods
Materials
PD was purchased from Xian kai lai Biological Engineering Co. Ltd. (Xian, China). Soy phospholipid was obtained from Shanghai Tywei Pharmaceutical Co. Ltd. (Shanghai, China). Methanol and acetonitrile were high performance liquid chromatography (HPLC). All other chemical reagents employed in the experiments were analytical purity grade. Water used in experiments was deionized.
Preparation of PPC
PPC was prepared by solvent evaporation method described as follows. In brief, the complex was prepared with the weight ratio of PD to soy phospholipid as 1:2, 1:3, and 1:4. Weighed amount of PD and soy phospholipid were dissolved into 20 mL tetrahydrofuran to obtain 5 mg/mL PD uniform pale yellow solution. The mixture was kept in a thermostatic water bath at 55°C for 1 h with magnetic stirring. At last, the eventual solution was evaporated by rotating decompression method. The complex was washed with n-hexane and dried under vacuum for 12 h to remove the remaining solvent. The product was stored in desiccator. The same ratio of PD and soy phospholipid was simply mixed to form physical mixture (PM) as control.
HPLC analysis
The concentration of PC samples was assayed by HPLC method. HPLC was equipped with Shimadzu LC-15C (shimadzu, Kyoto, Japan), SPD-15C UV-Spectrophotometric detector system. The analytical column was a COSMOSIL C18 column (4.6 mm × 250 mm, 5 μm; Nacalai Lnc, Kyoto, Japan) with temperature at 30°C. The mobile phase was comprised of water and acetonitrile (80:20, v/v) and delivered at a flow rate of 1.0 mL/min. The detection of PC was carried out at 322 nm and the injection volume was 20 μL.
PPC complexation rate
The complexation rate was calculated on the basis of the following equation:

Here, m1 is the amount of PD in PPC, and m2 is the amount of initial drug used.
Apparent solubility
To determine apparent solubility of PD and PPC, an excess amount of PD and PPC were added to 5 mL of deionized water. The samples were shaken for 24 h at room temperature and centrifuged at 1500 rpm for 20 min. The liquid supernatant was filtered with a 0.45 μm membrane filter. Filtrate was diluted with deionized water to reach appropriate concentration and measured at 322 nm by UV-spectrophotometer.
Fourier Transform Infrared (FT-IR) spectroscopic studies
The Fourier transform infrared (FT-IR) spectra of PPC, pure PD powder, phospholipids and physical mixture were performed on a Bruker Vector-22 FT-IR spectrometer. The spectra were recorded in the region of from 4000 to 500 cm-1 at room temperature. The samples were compressed into a KBr pellets.
Differential Scanning Calorimetry (DSC)
DSC curves of PD, phospholipids, and PPC were recorded using a differential scanning calorimeter. The thermal behavior was studied by heating 2.0 ± 0.2 mg of each individual sample in aluminium pan under nitrogen gas flow. The investigation was carried out over the temperature range 0-300°C at a heating rate of 10°C/min.
X-Ray Powder Diffraction (X-RPD)
The powder X-ray diffraction patterns (XRD) were measured by Philips X’Pert Pro Super Diffractometer Cu Kα radiation (λ=1.541874 Å), and the operating voltage and tube current were maintained at 40 kV and 40 mA, respectively. The scanning speed was 10°/min. The 2θ angle range was from 10 to 80° with a step size of 0.02° at room temperature.
In vitro release of PD
The solution of PD and PPC were prepared to investigate the dissolution profile using USP dissolution apparatus II method under sink conditions. Deionized water was used as the release medium. The paddle was rotated at 100 rpm and the temperature of the dissolution medium was set at 37 ± 0.5°C. Briefly, equivalent amount of PD for the two formulations were put into two swelled dialysis bag (molecular weight cut off 8000-14000 Da, USA) and the dialysis bag was placed into the release medium. At definite time intervals, 2 mL dissolution medium was withdrawn for sample analysis and replaced with an equal volume of pre-warmed fresh media. The PD samples were analyzed by UV spectrophotometer at 322 nm. All experiments were performed in triplicate.
Pharmacokinetic study in rats
All animal’s experimental protocols were approved by the Animal Care Committee of Anhui University of Chinese Medicine. Twelve healthy Sprague-Dawley (SD) rats were supplied from Experimental animal center of Anhui province (2011-002, Hefei, Anhui, China) and were housed in cages in appropriate temperature for 1 week before experimentation. Before dosed, the healthy rats were fasted overnight with free access to water and were divided randomly into two groups with half male and half female. Blood samples were collected into 1.5 mL centrifuge tubes at specified time intervals. Then the centrifuge tubes were centrifuged at 3000 rpm for 15 min to separate the plasma. The concentration of PD was estimated by HPLC method after acetonitrile protein precipitation. The software program 3P97 (Chinese Pharmacological Society, China) was employed to estimate the pharmacokinetic parameters. Data from different experimental groups were compared by using one-way analysis of variance (ANOVA) statistical analysis. All data were expressed as the means ± standard (S.D). The statistical analysis was made using the t-test. A p value less than 0.05 was considered statistically significant.
Results and Discussion
Preperation of PPC
PPC was prepared at weight ratio of phospholipids to drug as 2, 3 and 4. The final complexation rate of PPC were tested and calculated as 68%, 99.1% and 89.3%, respectively. Based upon the above result, it could be concluded 3:1 ratio of phospholipids to PD could get optimal complexation rate. When the ratio was more than 3 or less than 3, the complexation rate was decreased. So, the ratio of phospholipids to PD in our resultant formulation was set as 3:1. The complexation rate of PD in PPC with the optimal technology was 99.1%.
FT-IR spectra studies
FT-IR was used to investigate the possible interactions of PD and PPC, FTIR spectra shown in Figure 2 was characterized to investigate the possible interactions between PD and phospholipids in the phospholipid complex. The FTIR spectrum of PD (Figure 2a) exhibited the characteristic absorption bands were at 3480 cm-1, 1592 cm-1 and 1175 cm-1, which were attributed to -OH vibrations of the phenolic groups, C=C vibrations in the benzene ring and C-O vibrations of the phenolic groups, respectively. Moreover, the FTIR spectrum of phospholipids (Figure 2c) showed a broad hydroxyl stretching band at 3373 cm-1, C-H stretching band of long fatty acid chain at 2926 and 2855 cm-1, carbonyl stretching band at 1738 cm-1 in the fatty acid ester, P=O stretching band at 1240 cm-1, and N+(CH3)3 stretching at 970 cm-1.
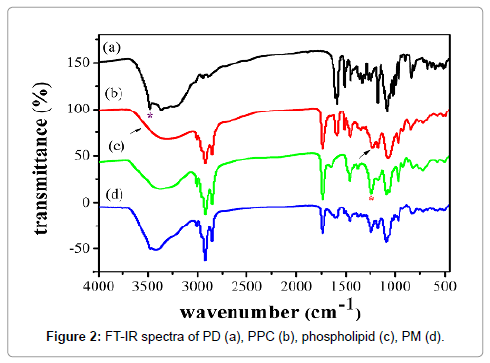
Figure 2: FT-IR spectra of PD (a), PPC (b), phospholipid (c), PM (d).
Compared to PD and phospholipids, the significant changes of the FTIR spectrum of PPC (Figure 2b) were observed. PPC showed that the absorption peak of phenolic -OH of PD was completely disappeared, and the P=O stretching band of phospholipids was almost disappeared. On the contrary, the spectrum of physical mixture (Figure 2d) was a simple summation of PD and phospholipids. These changes indicated there were some molecular interactions between the phospholipids molecule and these phenolic -OH groups in complex.
Differential Scanning Calorimetry (DSC)
DSC is a fast and reliable method to screen drug-excipient compatibility and provides maximum information about the possible interactions [26]. DSC thermograms of PD, phospholipids and PPC were shown in Figure 3. The DSC curve of PD (Figure 3a) exhibited a sharp exothermic peak at 219°C, which was attributed to its crystalline nature. There was no peak appeared in the thermogram of phospholipids (Figure 3c). The thermogram of PPC (Figure 3b) showed a broad and weak exothermic peak at 188°C, which was markedly different from the individual components of the complex. It was evident that the original peaks of PD disappeared from the thermogram of PPC complex and a new exothermic peak occurred in PPC compared to that of phospholipids. Generally, an interaction was concluded by the changes of exothermic peaks, which mainly included disappearance of peaks, appearance of new peaks, change in peak shape and its onset, peak temperature/melting point, and relative peak area or enthalpy [27]. Thus, the DSC date indicated the formation of the complex and the possible interaction between PD and phospholipids. The degree of the amorphization of PPC was significantly higher than PD, which resulted in the reduced crystallinity of the drug the increase of solubility.
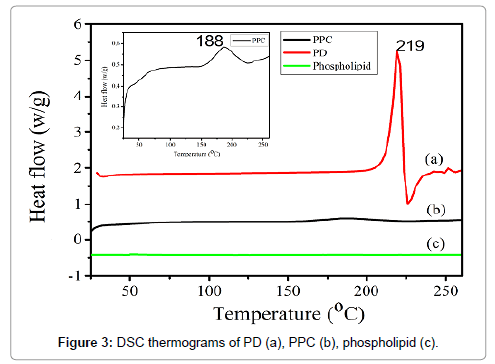
Figure 3: DSC thermograms of PD (a), PPC (b), phospholipid (c).
X-Ray Powder Diffraction (XRD)
To check the changes of crystal morphology of drug in complex, X-ray diffractometry is also a useful tool to study complex in the solid state. In the XRD, the peak position (angle of diffraction) is an indication of crystal structure and peak heights are the measures of sample crystallinity (crystallite size) in a diffractogram [28]. Figure 3 showed the X-ray powder diffraction patterns of PD and the complex. Sharp diffraction peaks near 10-40° for pure PD was shown in Figure 4a, which indicated the crystalline nature of PD. The peak shape suggested good crystallinity in PD powder. On the contrary, PPC shown in Figure 4b has no characteristic diffraction for PD. Compared with that of PD, there was a very few low-intensity crystalline peaks in the complex, indicating the vastly decreased crystallinity of PD and the formation of PPC. In the meantime, these information supports DSC studies which indicated that the crystallinity of drug was reduced in the complex. As the result, the XRD data showed an almost complete drug amorphization and/or complexation.
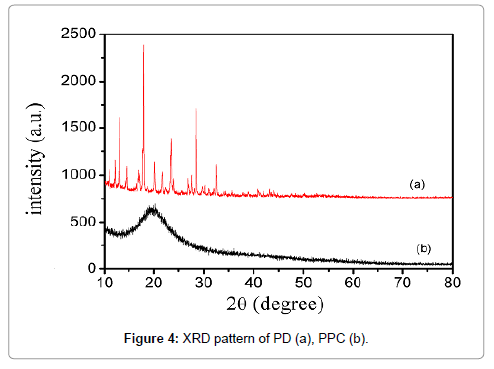
Figure 4: XRD pattern of PD (a), PPC (b).
Solubility study
The results of apparent solubility study were shown in Table 1, PPC exhibited better aqueous solubility than PD and the solubility of PPC in water increased 3.47-fold than pure PD. The low aqueous solubility of pure PD may be explained that the high crystallinity limited the solubility in water. On the contrary, PPC exhibited amorphous characteristics which were the primary causes in significantly improving aqueous solubility. Moreover, phospholipids could increase the solubility of the drug by the amphiphilic surfactant nature and the action of wetting and dispersion [29]. The better solubility may result in improved absorption in gastrointestinal tract and the enhancement of bioavailability.
| Samples |
Aqueous solubility (mg/L) |
| PD |
0.17 ± 0.01 |
| PPC |
0.59 ± 0.02 |
Table 1: Apparent solubility of PD and PPC (n=3).
In vitro release of PD
The in vitro release was conducted to evaluate the dissolution of PD and PPC in deionized water and the in vitro dissolution profiles were summarized in Figure 5. It can be seen that the cumulative release of PD and PPC were 63.84% and 80.36% in 8 h. Compared with PD, PPC significantly improved the cumulative release of the drug dissolution. The raising of accumulative dissolution rate of PPC mainly attributed to the drop of crystallinity. Instead of highly crystalline structure of the crude drug, the amorphous structure of PPC was conducive to dissolution. What’s more, being an amphiphilic surfactant, phospholipids increased the solubility of the drug by the action of wetting and dispersion.
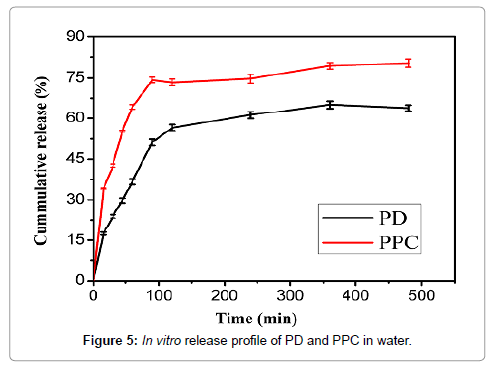
Figure 5: In vitro release profile of PD and PPC in water.
Pharmacokinetic study in rats
The in vivo evaluation was carried out to analyze the pharmacokinetics by oral administration of PD and PPC in rats. The plasma concentration-time profiles was presented in Figure 6. The pharmacokinetic parameters were calculated by the employ of the software 3p97. Meanwhile, the major pharmacokinetic parameters are shown in Table 2. The main pharmacokinetic parameters of PPC were significantly improved compared to PD group. The Cmax of PD was 1.21 μg/mL with Tmax of about 90 min after oral administration. However, the Cmax of PPC was 2.60 μg/mL with Tmax of about 150 min. The results showed that the Cmax of PPC were markedly higher than PD with longer Tmax, which inferred the considerable enhancement in oral bioavailability of PPC. The AUC0-∞ of PPC was 748.2 μg·min/mL, which was approximately 2.2 folds than PD (340.6 μg·min/mL). The enhancement of the relative bioavailability may be attributed to the following reason: As an indispensable constituent of cell membrane, phospholipids play a key role in keeping cell membrane fluidity, which makes it possible that the complex cross cell membranes without disturbing the cellular lipid bilayers. Identically, the phospholipid complex are liable to transported from a hydrophilic environment to a lipophilic environment of the enterocyte cell membrane, then into the cell and lastly into the systemic blood circulation [30]. Before drugs permeate the GI (gastric and intestinal) tract membranes to reach the systemic circulation, the dissolving ability of drugs in gastric and/or intestinal fluids is an very important limiting factor [31]. This study had shown that the PPC improve the dissolution behavior of PD in water. As a result, the PD absorption in GI tract was maybe increased. In all, these changes were mainly due to the raise of lipophilicity and solubility of PPC.
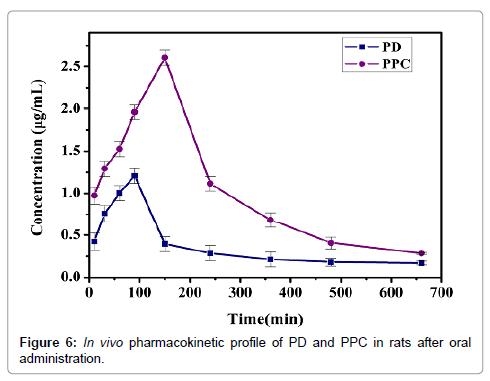
Figure 6: In vivo pharmacokinetic profile of PD and PPC in rats after oral administration.
| Parameters |
PPC |
PD |
| Tmax (min) |
150 |
90 |
| Cmax (µg/mL) |
2.60 |
1.21 |
| AUC0-t (µg·min/mL) |
661.7 |
237.0 |
| AUC0-8 (µg·min/mL) |
748.2 |
340.6 |
| Cl(µg/mL) |
0.40 |
0.88 |
Table 2: Pharmacokinetic parameters of PD and PPC solution in rats after oral administration (mean ± S.D., n=6).
Conclusion
In this research, PPC was successfully prepared with high complexation rate by the solvent evaporation method. FTIR, DSC and XRD characterizations of phospholipid complex confirmed that PPC was formed by the molecular interactions between PD and phospholipids, such as hydrogen bonds or van der Waals force. PPC exhibited better solubility and significant increase of in vitro dissolution compared to PD. The in vivo study indicated that the oral bioavailability (Cmax and AUC0-t) of PPC was markedly improved compared to PD. And the absorption mechanism of PPC through small intestine and in vivo therapeutic evaluation should be further studied. In general, PPC would be a promising formulation for PD to further exploration.
Acknowledgements
We appreciate the financial support provided by Anhui University of Traditional Chinese Medicine. This research was supported by the National Natural Science Foundation of China (Grants 51303006) and the Provincial Natural Science Foundation (1408085MH196, KJ2012ZD09) of Anhui Province.
Declaration of Interest
The authors report no conflict of interest.
18765
References
- Ince S, Acaroz DA, Neuwirth O, Demirel HH, Denk B, et al. (2014) Protective effect of polydatin, a natural precursor of resveratrol, against cisplatin-induced toxicity in rats. Food and Chemical Toxicology 72: 147-153.
- Yiu CY, Chen SY, Yang TH, Chang CJ, Yeh DB, et al. (2014) Inhibition of Epstein-Barr virus lytic cycle by an ethyl acetate subfraction separated from Polygonum cuspidatum root and its major component, emodin. Molecules 19: 1258-1272.
- Leu YL, Hwang TL, Hu JW, Fang JY (2008) Anthraquinones from Polygonum cuspidatum as tyrosinase inhibitors for dermal use. Phytotherapy Research 22: 552-556.
- Zhang C, Wang X, Zhang X, Zhang Y, Xiao H, et al. (2009) Bioassayâ€ÂÂÂÃÂguided separation of citreorosein and other oestrogenic compounds From Polygonum cuspidatum. Phytotherapy research 23: 740-741.
- Kirino A, Takasuka Y, Nishi A, Kawabe S, Yamashita H, et al. (2012) Analysis and functionality of major polyphenolic components of Polygonum cuspidatum (itadori). Journal of nutritional science and vitaminology, 58: 278-286.
- Du QH, Peng C, Zhang H (2013) Polydatin: a review of pharmacology and pharmacokinetics. Pharmaceutical biology 51: 1347-1354.
- Du J, Sun LN, Xing WW, Huang BK, Jia M, et al. (2009) Lipid-lowering effects of polydatin from Polygonum cuspidatum in hyperlipidemic hamsters. Phytomedicine 16: 652-658.
- Zhang Y, Zhuang Z, Meng Q, Jiao Y, Xu J, et al. (2014) Polydatin inhibits growth of lung cancer cells by inducing apoptosis and causing cell cycle arrest. Oncology letters 7: 295-301.
- Liu LT, Guo G, Wu M, Zhang WG (2012) The progress of the research on cardio-vascular effects and acting mechanism of polydatin. Chinese journal of integrative medicine 18: 714-719.
- Kerem, Z, Bilkis I, Flaishman MA (2006) Antioxidant Activity and Inhibition α-Glucosidase by trans-Resveratrol, Piceid, and a Novel trans-Stilbene from the Roots of IsraeliRumex bucephalophorus. L. J J Agric. Food Chem. 54: 1243-1247.
- Wang X, Guan Q, Chen W, Hu X, Li L (2015) Novel nanoliposomal delivery system for polydatin: preparation, characterization, and in vivo evaluation. Drug design, development and therapy 9: 1805.
- Wang H, Cui Y, Fu Q, Deng B, Li G, Yang J, et al. (2015) A phospholipid complex to improve the oral bioavailability of flavonoids. Drug development and industrial pharmacy 41: 1693-1703.
- Singh D, SM Rawat M, Semalty A, Semalty M (2012) Quercetin-phospholipid complex: an amorphous pharmaceutical system in herbal drug delivery. Current drug discovery technologies 9: 17-24.
- Rabinovich-Guilatt L, Dubernet C, Gaudin K, Lambert G, Couvreur P, et al. (2005) Phospholipid hydrolysis in a pharmaceutical emulsion assessed by physicochemical parameters and a new analytical method. European journal of pharmaceutics and biopharmaceutics 61: 69-76.
- Sha X, Guo J, Chen Y, Fang X (2012) Effect of phospholipid composition on pharmacokinetics and biodistribution of epirubicin liposomes. Journal of liposome research 22: 80-88.
- García-Moreno PJ, Horn AF, Jacobsen C (2014) Influence of casein–phospholipid combinations as emulsifier on the physical and oxidative stability of fish oil-in-water emulsions. Journal of agricultural and food chemistry 62: 1142-1152.
- Kidd PM (2009) Bioavailability and activity of phytosome complexes from botanical polyphenols: the silymarin, curcumin, green tea, and grape seed extracts. Altern Med Rev 14: 226-46.
- Chen ZP, Sun J, Chen HX, Xiao YY, Liu D, et al. (2010) Comparative pharmacokinetics and bioavailability studies of quercetin, kaempferol and isorhamnetin after oral administration of Ginkgo biloba extracts, Ginkgo biloba extract phospholipid complexes and Ginkgo biloba extract solid dispersions in rats. Fitoterapia 81: 1045-1052.
- Zhang K, Gu L, Chen J, Zhang Y, Jiang Y, et al. (2015) Preparation and evaluation of kaempferol–phospholipid complex for pharmacokinetics and bioavailability in SD rats. Journal of pharmaceutical and biomedical analysis 114: 168-175.
- Khan J, Alexander A, Saraf S, Saraf S (2014) Luteolin–phospholipid complex: preparation, characterization and biological evaluation. Journal of Pharmacy and Pharmacology 66: 1451-1462.
- Sikarwar MS, Sharma S, Jain AK, Parial SD (2008) Preparation, characterization and evaluation of marsupsin–phospholipid complex. AAPS PharmSciTech 9: 129-137.
- Jin X, Zhang ZH, Sun E, Qian Q, Tan XB, et al. (2012) Preparation of a nanoscale baohuoside I-phospholipid complex and determination of its absorption: in vivo and in vitro evaluations. International journal of nanomedicine 7: 4907.
- Song Y, Zhuang J, Guo J, Xiao Y, Ping Q (2008) Preparation and properties of a silybin-phospholipid complex. Die Pharmazie-An International Journal of Pharmaceutical Sciences 63: 35-42.
- Maiti K, Mukherjee K, Gantait A, Saha BP, Mukherjee PK (2007) Curcumin–phospholipid complex: preparation, therapeutic evaluation and pharmacokinetic study in rats. International journal of pharmaceutics 330: 155-163.
- Jena SK, Singh C, Dora CP, Suresh S (2014) Development of tamoxifen-phospholipid complex: novel approach for improving solubility and bioavailability. International journal of pharmaceutics 473: 1-9.
- Li Y, Jin W, Yan H, Liu H, Wang C (2013) Development of intravenous lipid emulsion of vinorelbine based on drug–phospholipid complex technique. International journal of pharmaceutics 454: 472-477.
- Guo B, Liu H, Li Y, Zhao J, Yang D, et al. (2014) Application of phospholipid complex technique to improve the dissolution and pharmacokinetic of probucol by solvent-evaporation and co-grinding methods. International journal of pharmaceutics 474: 50-56.
- Semalty A (2014) Cyclodextrin and phospholipid complexation in solubility and dissolution enhancement: a critical and meta-analysis. J. Expert Opin Drug Deliv 11: 1255-1272.
- Yanyu X, Yunmei S, Zhipeng C, Qineng P (2006) The preparation of silybin–phospholipid complex and the study on its pharmacokinetics in rats. International journal of pharmaceutics 307: 77-82.
- Sharma S, Roy RK (2010) Phytosomes: an emerging technology. Int J Pharm Res Dev 2: 1-5.
- Jiang Q, Yang X, Du P, Zhang H, Zhang T (2016) Dual strategies to improve oral bioavailability of oleanolic acid: Enhancing water-solubility, permeability and inhibiting cytochrome P450 isozymes. European Journal of Pharmaceutics and Biopharmaceutics 99: 65-72.


