Rayya A. Almarwani*, Fahad A. Alsayed, Ehab S. Showki, Khalid O Alahmadi, Hesham Aldhalaan
Department of Pediatric Neurology, King Faisal Specialist Hospital and Research Center, Saudi Arabia
Corresponding Author:
Dr. Rayya Almarwani
Department of Pediatric Neurology, King Faisal Specialist Hospital and Research Center, Saudi Arabia
Tel: 00966508878467
E-mail: rayya.aljohani@gmail.com
Rec Date: October 10, 2018; Acc Date: November 05, 2018; Pub Date: November 09, 2018
Citation: Almarwani RA, Alsayed FA, Showki ES, Alahmadi KO, Aldhalaan H (2018) Progressive Cavitating Leukoencephalopathy in an Infant Baby: A Case Report. J Neurol Neurosci Vol.9 No.6:274. doi:10.21767/2171-6625.1000274.
Keywords
Genetic mutation; Infant; Leukodystrophy; Mitochondria dysfunction syndrome; Saudi Arabia
Introduction
Cavitating leukoencephalopathy is part of a group of disorders that is characterized by degeneration of the white matter in the brain. The disease in neonates has severe neurological and systematic manifestations, including seizures, hypotonia, failure to thrive, lactic acidosis, and respiratory failure. Multiple genetic mutations have been identified such as the mutation in the BOLA3 gene (613183) on chromosome 2p13; MMDS3 (615330) is caused by a mutation in the IBA57 gene (615316) on chromosome 1q42 and MMDS4 (616370) is caused by a mutation in the ISCA2 gene (615317) on chromosome 14q242. The above-mentioned genes are responsible for the regulation of the iron-sulfur (Fe-S) biosynthesis pathway. Various proteins contain Fe-S, including mitochondrial respiratory chain (MRC) complexes I, II, and III, and lipoic acid synthase (LIAS). IBA57 proteins are crucial components of the mitochondrial iron-sulfur cluster assembly machinery [1,2]. It has been shown that these proteins are specifically required for the generation of [4Fe-4S] clusters and their dedicated insertion into mitochondrial Apo proteins. Diseases arising from mutations in these genes are expected to elicit defects in mitochondrial respiration and in lipoic aciddependent proteins [3]. The present report presents the case of a 7-month-old boy who died of MMDS with progressive cavitating leukoencephalopathy, and we confirmed that he and his parents had the IBA57 mutation. This is the first reported case of leukoencephalopathy in Saudi Arabia and in the Gulf region.
Case Report
At the outpatient pediatric neurology clinic of King Faisal specialist Hospital, Riyadh city, Saudi Arabia, a 5-month-old boy presented with excessive frequent crying since birth. The baby was full term, weighed 2000 grams at birth, and was delivered by normal spontaneous vaginal delivery after an uneventful pregnancy. He was the second product of a firstdegree consanguineous marriage. At 3 months of age, the baby became more irritable and had decreased oral intake. He also had a history of abnormal movement, described as tonic spasm of the upper limbs, for a few seconds, delayed developmental milestones, and regression of the achieved milestones. The baby’s past medical history was significant for recurrent spiking fever secondary to urinary tract infection with positive urine cultures for extended-spectrum betalactamase- producing Escherichia coli. His older sister, who died at 14 months of age, had similar symptoms and presentation. Physical examination showed a crying baby with an inability to fix his eyes or follow objects. The anterior fontanel was open with a head circumference of 46 cm at the 25th percentile. The weight and height of the child was within the 10th percentile. No organomegaly was found. Neurological examination of the patient showed spastic tone (greater in the upper limbs), exaggerated limbs and gags reflexes, bilateral clonus, and truncal hypertonia. Eye examination was positive for horizontal eye nystagmus and optic nerve atrophy. No neurocutaneous stigmata were detected. Laboratory work-up showed normal complete blood count, liver function tests, and thyroid function results. The blood lactic acid level was slightly elevated, but plasma amino acid analysis was normal. Magnetic resonance imaging (MRI) and magnetic resonance spectroscopic (MRS) showed cystic leuckodytrophy along course of cortico-spinal tracts (Figure 1).
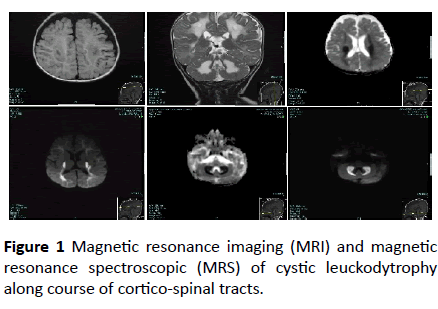
Figure 1: Magnetic resonance imaging (MRI) and magnetic resonance spectroscopic (MRS) of cystic leuckodytrophy along course of cortico-spinal tracts.
Diffusion weighted imaging and apparent diffusion coefficient (DWI/ADC) map showed significant diffusion restriction in bilateral centrum semi-ovale, periventricular white matter and posterior limbs of internal capsule along course of cortico-spinal tracts and bilateral dentate nuclei. T2 sequence showed hyperintense signal that demonstrate drop in signal in FLAIR sequence consistence with vacuolization and cystic degeneration (Figure 2).
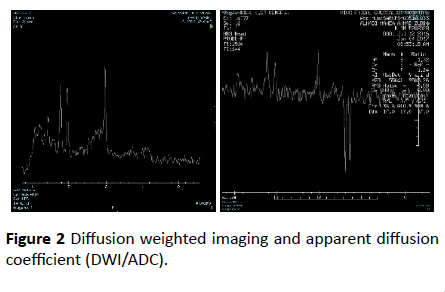
Figure 2 Diffusion weighted imaging and apparent diffusion coefficient (DWI/ADC).
MRI spectroscope showed very high lactate peak in 1.3 ppm in TE 144 consistence with lactic acidosis, and the low metabolic peaks in TE 35 corresponding with loss of neuronal tissue.
Comprehensive Epilepsy panel (NextGen Sequencing panel 165 genes +mtDNA) detected IBA57, c.316a>G, P. Thr106Ala homozygous autosomal recessive condition.
Unfortunately, the patient died 2 months later as a result of acute respiratory failure secondary to infection. The final diagnosis of the case was MMDS with progressive cavitating leukoencephalopathy. The IBA57 homozygous mutation of both parents was detected with the heterozygous of the same gene.
Discussion
There are numerous genes that cause mitochondrial disorders and clinically and genetically heterogeneous disorders whose etiology often remains unsolved. Of these genes, IBA57 proteins are crucial components of the mitochondrial iron-sulfur cluster assembly machinery. Its mutation, however, was implicated in MMDS in infancy [4].
The reported data in 2013 have demonstrated two siblings from consanguineous Moroccan origin parents died perinatally, with a condition characterized by generalized hypotonia, respiratory insufficiency, arthrogryposis, microcephaly, congenital brain malformations and hyperglycinemia. Homozygosity mapping identified of these two babies showed IBA57 homozygous mutation located in the largest homozygous region on chromosome 1 as a culprit candidate gene [5].
Much more recently, two children patients with a cavitating leukodystrophy secondary to mutations in the iron cluster assembly gene IBA57 were reported in 2017 [6]. The first patient was a female born to consanguineous parents. She developed normally until 6 months after which she had a rapid regression in motor and visual skills. At 11 months’ examination revealed irritability, evolving quadriparesis, poor vision and sluggish pupillary reactions. MRI of this female patient showed extensive bilateral leukoencephalopathy with the involvement of the corpus callosum and middle cerebellar peduncles and large cavitary areas were found in the deep white matter. Furthermore, homozygous mutations in the IBA57 gene were identified in the patient [6]. The second patient was a male born to no consanguineous parents. In this patient, developmental concerns at 6 months were noticed, particularly concerning vision. Examination at 17 months showed a mild hypertonia of all limbs, poor vision, and optic atrophy. He made developmental progress until the age of 7 where he has severe visual impairment, four limbed spasticity and epilepsy. MRI at 17 months demonstrated a diffuse leukoencephalopathy with large cavitated areas. Progression of cavitation, a small corpus callosum but with some improvement in myelination was detected at the age of 6 years. The molecular investigation demonstrated compound heterozygous mutations in IBA57 gene [6].
The clinical course of this disease varies from rapid deterioration to prolonged periods of stability. The present newly born patient started to develop the neurological symptom with blindness and spasticity following. Like other reported cases, the present case showed abnormal findings on MRI including cavitation and cystic leuckodytrophy and IBA57 gene mutation on homozygosity mapping. Familial occurrence and consanguinity appeared to play an important role in this disease and suggest autosomal recessive inheritance of this distinct entity. In the present and other reported cases consanguinity has been a matter, and positive family history has been recorded in the present case report.
Conclusion
In conclusion, the disease is a very rare mutation, but it is rapidly progressive and fatal disorder. The disease is primarily a leukoencephalopathy and the homozygosity of IBA57 gene is the key concern in mitochondrial dysfunction and progressive cavitating leukoencephalopathy with progression to respiratory failure and death. According to available data, this infant case of MMDS with cavitating leukoencephalopathy is the first to be reported in Saudi Arabia as well as in the Gulf region.
23734
References
- Perminder S, Matcheri S (2010) Secondary schizophrenia. Cambridge University Press, USA.
- Seyda A, Newbold RF, Hudson TJ, Verner A, MacKay N, et al. (2001) A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet 68: 386-396.
- Stehling O, Wilbrecht C, Lill R (2014) Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 100: 61-77.
- Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsasser HP, et al. (2012) The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Molec Biol Cell 23: 1157-1166.
- Bolar NL, Vanlander AV, Wilbrecht C, Van der N, Smet J, et al. (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet 22: 2590-2602.
- Green LMC, Berry I, Warren D, Livingston JH (2017) Mutaton in the iron sulpher cluster assemply gene IBA57 cause a cavitating leukodystrophy associated with a variable phenotype. European Journal of Paediatric Neurology 21: e50.

