Keywords
Cerebral blood flow; Mice; Neuroprotection; Nitric oxide synthase; NMDA; Stroke; Vasodilation
Introduction
After the onset of ischemic brain injury, a wide array of factors adds up to a total pathophysiology that includes but is not limited to excitotoxicity, inflammation, free radical damage, and cell death. Excitotoxicity and free radical generation are considered early events in stroke and have been implicated as major players in stroke pathophysiology [1]. After the onset of ischemia, there is a fall in pH, a depletion of ATP, and then a failure of the Na+/K+ pump, resulting in membrane depolarization. This leads to a rapid accumulation of intracellular glutamate to a toxic level that then leads to an increase in the intracellular level of Ca2+. Thus, the elevation of intracellular Ca2+ resulting from the activation of the N-methyl- D-aspartate (NMDA) receptor activates Ca2+-dependent nitric oxide (NO) synthases (NOS) and NO production [2,3]. NO has been reported to play critical physiological roles such as vasodilation, cytotoxicity, prevention of invading pathogens, and pathogenesis of inflammatory diseases [4-7]. Further, free radicals generated during pathologic conditions interact with NO and form the highly toxic peroxynitrite [8-10].
Arginase (Arg) is the final enzyme in the urea cycle and is essential in the route for the disposal of excess nitrogen resulting from amino acid and nucleotide metabolism [11]. It has been reported that intravenous administration of Arg exerts significant effects on circulation, which suggests the potential role of this enzyme in regulating vascular function [12]. In addition, Arg competes with NOS to use L-arginine as a substrate, and subsequently regulates NOS activity for both constitutive and inducible NOS isoforms [13]. In the process, two different metabolic pathways involved are NOS reacting with L-arginine, resulting in citrulline and nitric oxide (NO) or Arg reacting with L-arginine, leading to the production of ornithine and urea. Thus far, two isoforms of Arg (ArgI and ArgII) have been found. ArgI, predominantly expressed in the liver, has been found to be present throughout the body, whereas ArgII, a mitochondrial enzyme, is expressed in the kidneys, brain, liver, small intestine, mammary glands, and macrophages [7,14,15]. However, it is believed that both isoforms are induced during different pathophysiological conditions [10,16]. Studies have shown that down-regulation of ArgII leads to renal apoptosis, whereas overexpression inhibits renal apoptosis during inorganic mercury exposure [17]. Recent studies have suggested that ArgII is a major isoenzyme in the endothelial cells (ECs) of humans and other species, and an important regulator of NO production [15].
Stroke is considered a leading cause of death in the United States and worldwide. Its treatment has been eluding scientists for many decades, with no current ideal therapeutic alternative. Although a wide array of possible approaches have been postulated, most fail at clinical trials. Therefore, there is an urgent need to look into the endogenous intracellular events that could be utilized as therapeutic targets or agents in minimizing/preventing ischemic brain injury and excitotoxicity. Accumulating evidence shows that Arg plays a critical role in modulating NO and NOS, with NOS considered a potent vasoprotective molecule that regulates vascular tone and controls vascular integrity [18]. In manipulating NOS isoforms, Arg and NO would be useful in developing therapies to modulate the response of an organism to cerebral ischemic injury. Here, we hypothesize that the genetic deletion of ArgII, an important component for normal functioning of blood vessels, causes increased availability of L-arginine as a substrate to NOS, thereby increased NO concentration, which leads to reactive oxygen/nitrogen species generation and augmented brain damage. To test our hypothesis, we subjected ArgII-/- mice to permanent distal middle cerebral artery occlusion (pMCAO) and the findings were further tested in a more specific NMDA-induced acute excitotoxicity model.
Material and Methods
Animals
Male C57BL/6 wildtype (WT) mice weighing 20-25 g were purchased from Charles River Laboratories Inc. (Wilmington, MA). Age- and weight-matched ArgII-/- mice were provided by Dr. Dan E. Berkowitz, MBBCh (Johns Hopkins University, Baltimore, MD). Animals were housed in our institutional animal facility and had access to food and water ad libitum under controlled conditions. All procedures and protocols were approved by the Institutional Animal Care and Use Committee of The Johns Hopkins University School of Medicine. Investigators were blinded to the experimental groups.
Permanent distal middle cerebral artery occlusion (pMCAO)
The distal section of the MCA was occluded as originally described by us and others [19,20]. Briefly, WT (n=8) and ArgII-/- (n=10) mice were anesthetized with continuous 1% halothane, and a 1.0-cm vertical skin incision was drawn between the right eye and ear. The temporal muscle was moved aside to expose the underlying temporal bone. Under a surgical microscope, a 2.0-mm burr hole was drilled just over the MCA, visible through the temporal bone. Using a bipolar coagulator, the main trunk of the distal part of the MCA was directly occluded, and complete interruption of blood flow at the occlusion site was confirmed by the severance cerebral blood flow (CBF) at the MCA site. Core body temperature was maintained at 37.0 ± 0.5°C during the procedures with the help of an automated heating blanket. After the procedures, mice were placed in an automated temperature- and humidity-regulated recovery chamber for 2 h before being moved to a clean cage. The animals not circling toward the paretic side after the onset of ischemia and those that developed subarachnoid hemorrhage were not included in the study. A successful occlusion was confirmed only after placing the laser-Doppler probe above the temporal ridge to establish that relative CBF (rCBF) into the region was terminated.
Assessment of neurological deficit score (NDS) in pMCAO
To evaluate the neurological deficits caused by this model, a 28-point score pattern was used, as reported earlier. Seven days after the pMCAO procedure, an experimenter blinded to genotype scored all mice for neurological deficits. The tests included body symmetry, gait, climbing, circling behavior, front limb symmetry, compulsory circling, and whisker response. Each test was graded from 0 to 4, establishing a maximum NDS of 28. Immediately after the evaluation, the mice were sacrificed for infarct volume analysis.
Blood gas and CBF measurements
A separate cohort was used to investigate the effect of pMCAO on blood gases and CBF in WT and ArgII-/-. Briefly, under an operating microscope, mice (n=5) were placed supine and an incision was made to expose the femoral artery. A PE-10 femoral artery catheter (Intramedic; BD Diagnostic Systems, Sparks, MD) attached to a 1-mL syringe was introduced into the artery and secured with a silk suture. Blood samples were drawn 60 min before and 90 min after pMCAO and were analyzed by blood gas analysis (Rapidlab 248; Chiron Diagnostic Corporation, Norwood, MA) for pH, PaO2, and PaCO2. Simultaneously, mice were recorded for rCBF during the entire procedure from 15 min prior to pMCAO to 120 min after pMCAO.
NMDA-induced acute excitotoxicity
To determine the role of ArgII on NMDA toxicity, WT and ArgII-/- mice (n=7/group) were given a single injection of 15 nmol NMDA (in 0.3 μL) or vehicle in the right striatum, as described [21]. After 5 min, the needle was slowly retracted, the wound was sutured, and the animals were transferred to an automated temperature- and humidity-regulated recovery chamber for 2 h before moving them to a clean cage. Throughout the experimental procedure, the rectal temperature of the mice was monitored and maintained at 37.0 ± 0.5ºC.
Excitotoxic lesion volume assessment
At 48 h after NMDA injection, weight and rectal temperature were recorded. The mice were deeply anesthetized with pentobarbital sodium (65 mg/kg) and were then perfused transcardially with cold phosphate-buffered saline (PBS) followed by fixation with 4% paraformaldehyde (pH=7.2) in PBS. Brains were removed quickly, post-fixed overnight, and equilibrated in 30% sucrose. The snap-frozen brains were cut on a cryostat and 25 μm sequential brain sections were stained with Cresyl violet to estimate lesion volume [21].
Statistical analysis
Data are expressed as mean ± SEM and were analyzed by unpaired t test. NDS was analyzed by the non-parametric Mann Whitney test. Statistical significance was set at p<0.05.
Results
Protective role of ArgII against neurologic deficits induced by pMCAO
Wildtype and ArgII-/- mice subjected to pMCAO, were monitored over a period of 7 days for any severe complications and, before sacrificing, were evaluated for NDS. There was no significant weight loss observed in either WT or ArgII-/- mice after stroke (data not shown). ArgII-/- mice were observed to have significantly (p=0.0019) higher neurologic outcomes (19.5 ± 1.5) compared to WT mice (12.2 ± 1.3). WT mice performing better and having a lower NDS than ArgII-/- mice reflect the role of ArgII in the recovery of mice from delayed ischemic insults (Figure 1A).
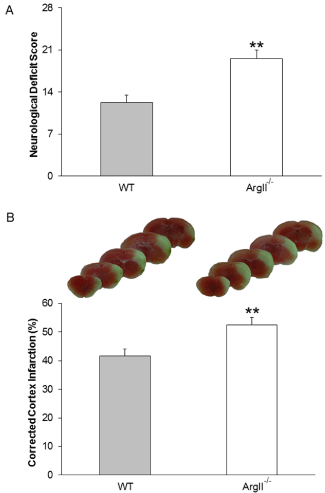
Figure 1: Deletion of ArgII leads to increased neurological deficit score and infarct volume. WT (n=8) and ArgII-/- (n=10) mice were subjected to permanent distal middle cerebral artery occlusion (pMCAO) and were allowed to survive for 7 days. On day 7 mice were sacrificed immediately after being tested for the NDS and brain infarction was analyzed after triphenyltetrazolium chloride staining. (A) As an indicator of neurological dysfunction, assessment on day 7 after ischemia show significantly larger neurological deficit scores in ArgII-/- mice than in WT mice. (B) Representative macrographs showing the slices of brain infarction from WT (left) and ArgII-/- (right) mice. (C) Histogram shows the percent corrected cortex infarct volume of WT and ArgII-/- mice. The infarct size was significantly larger in ArgII-/- than in WT mice. Data represented as mean ± S.E.M; *p=0.0019, **p=0.0079.
ArgII deletion exacerbates brain infarction induced by pMCAO
Immediately after the functional outcomes assessment, mice were sacrificed. Brain sections stained with triphenyl tetrazolium chloride (TTC) and analysis thereafter revealed that ArgII-/- mice suffered with significantly (p=0.0079) higher infarct volumes (52.4 ± 2.7 mm3) compared to their WT counterparts (41.6 ± 2.4 mm3; Figure 1B). WT mice having smaller infarction volume indicate that ArgII plays a substantial protective role in ischemic events.
Physiological parameters and CBF
In a separate cohort of mice involving WT and ArgII-/- mice, no differences in blood pH, PaO2, or PaCO2 were observed between the two groups before or after pMCAO (Table 1). In this same group of mice, CBF was measured continuously from 15 min prior to occlusion through 120 min after. CBF in ArgII-/- mice was observed to be non-significantly lower at occlusion time, followed by a further non-significant drop at later time points compared with WT mice. WT mice had a relatively stable CBF disruption, showing that ArgII deletion may have partial adverse effects on CBF (Figure 2).
| Parameters |
60min before pMCAO |
90min after pMCAO |
| WT |
| pH |
7.31 ± 0.04 |
7.26 ± 0.06 |
| PaCO2 |
45.4 ± 1.6 |
45.2 ± 1.7 |
| PaO2 |
139 ± 5.8 |
130 ± 6.5 |
| ArgII-/- |
| pH |
7.32 ± 0.02 |
7.26 ± 0.01 |
| PaCO2 |
43.8 ± 1.7 |
44.2 ± 1.5 |
| PaO2 |
132 ± 4.5 |
131 ± 3.2 |
Table 1 Physiologic parameters of WT and ArgII-/- mice before and after pMCAO.
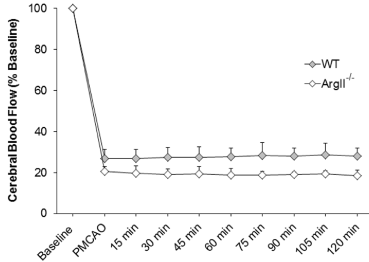
Figure 2:ArgII deletion attenuates relative cerebral blood flow rCBF. rCBF was recorded at baseline, at induction of ischemia, and at 15-min intervals during ischemia until 120 min in WT and ArgII-/- mice (n=5, each group). ArgII-/- exhibited a non-significant decrease in CBF. Changes in CBF were recorded as a percent of baseline. Data are represented as mean ± S.E.M.
Augmentation of NMDA-induced neurotoxicity in ArgII-/- mice
To further determine the contribution of ArgII to the pathology of stroke, we examined whether NMDA-induced acute excitotoxicity would be aggravated in ArgII-/- mice. Analyses of the Cresyl violet-stained brain sections revealed a significant (p=0.035) increase in the NMDA-induced lesion volume in the ArgII-/- mice than in the WT mice (Figure 3).
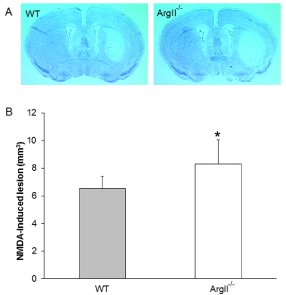
Figure 3: ArgII deletion increases NMDA-induced neurotoxicity. WT and ArgII-/- mice (n=7, each group) were given a single stereotaxic injection of 15 nmol NMDA in the striatum and sacrificed 48 h later. Brain sections were stained with Cresyl violet to analyze the brain lesions. (A) Representative macrographs of coronal sections of the WT (left panel) and ArgII-/- (right panel) mice brain after intrastriatal injection with 15 nmol NMDA. The brain sections from the ArgII-/- mise appears to show greater lesion volume. (B) Analysis of the brain sections show that the ArgII-/- mice were more vulnerable to the NMDAinduced neurotoxicity than were the WT mice. Values are represented as means ± S.E.M.; *p=0.035, when compared with WT group.
Discussion
In the present study, we evaluated the role of ArgII in cerebral ischemia and excitotoxic brain injury by using two complementary preclinical models. The pMCAO model provides an understanding of the delayed ischemic events that take place after the induction of ischemia; on the other hand, NMDA-induced excitotoxicity highlights the consequences of an acute excitotoxic insult that occurs immediately following stroke. Excitotoxicity and the generation of reactive oxygen species (ROS) are among the early systemic cascade of ischemic events leading to brain damage. Here, we have shown that ArgII is an important factor in controlling the damage induced by two different models of acute brain insults. ArgII-/- mice were observed to have higher infarction volume and higher NDS when subjected to pMCAO. Similarly, ArgII-/- mice were also observed to suffer higher damages caused by NMDA injections compared to WT mice. Together, the data suggest that deletion of ArgII renders mice more susceptible to insults and therefore reveals an important role of ArgII in cerebral ischemia and excitotoxicity.
The findings that ArgII-/- mice suffer from higher infarction volumes and neurologic deficits in a pMCAO-induced model serves as an indication that ArgII is a highly important enzyme and has a protective role during ischemic events. Thus far, the role of ArgII, using the ArgII-/- mice, has not been studied in cerebral ischemia and ours is the first study to demonstrate the protective effects of ArgII in the brain. There is emerging evidence available in the literature about the critical role of ArgII in regulating NO production [18,22]. NOS contain binding sites for L-arginine, reduced nicotinamide adenine dinucleotide phosphate, and Ca2+/calmodulin. It has been reported that certain conditions such as shear stress results in a Ca2+ influx into the endothelial cell and subsequently facilitates the generation of free radicals [8,23]. Nitric oxide plays vital role in vasodilation and previous reports have directly correlated ArgII with the modulation of NO production and, consequently, endothelial cell function [10,15,24]. Of interest, with the given experimental protocol, our results revealed a trend to attenuate CBF in ArgII-/- mince compared with WT mice, suggesting that deletion of ArgII may have deleterious effects on CBF. Our finding is supported by another study where researchers showed that administration of Arg increased vasodilation in mice [25].
Moreover, the inhibition of Arg has been shown to stimulate NO and peroxynitrite synthesis in endothelial cells [10]. Similarly, in our study, the absence of ArgII might stimulate NO synthesis, which then interacts with superoxides and participates in the generation of highly toxic peroxynitrite [8,9]. Furthermore, it should be noted that under certain conditions (e.g. aging, diabetes, hypertension, and atherosclerosis), Arg upregulation leads to an Arg/NOS imbalance and decreased NO production, which can contribute to endothelial dysfunction [15,26]. A previous report using rat model of an anoxia/reperfusion model shows that the reperfusion group had increased Arg levels; however, in the anoxia-only group, the Arg levels remain unchanged [27]. The authors concluded that this increase in Arg level attenuated NO, which would subsequently limit oxidative stress. Our study does not contradict this previous study because in our study, we used ArgII-/- mice; however, one cannot neglect the potential compensatory pathways and the fact that ArgI could also be a factor in dictating outcomes. Furthermore, our pMCAO model is likely more related to an anoxia model (low blood flow and no reperfusion) in which Swamy et. al. [27] found no changes in Arg level. Therefore, these studies differ in the use of models targeting either total Arg or ArgII only and consequently, different outcomes were observed.
Our findings in ArgII-/- were further extended in an acute model of excitotoxicity to determine whether our observations in an ischemic model could be potentially due to excitotoxicity and oxidative stress. Glutamate and NMDA, and their receptors, are vital for normal neurotransmission and physiological processes. However, the pharmacological hyperactivation of these receptors by toxins results in hyperactivity, leading to a pathological condition known as excitotoxicity, which is one of the early events in propagating neurological damage in stroke [28-33]. The increased level of Ca2+ in excitotoxicity activates NOS and leads to the generation of NO and ROS, which causes oxidative stress. Moreover, it has been proposed that NO mediates glutamate toxicity [34]. Thus, in the absence of ArgII, L-arginine is readily available to NOS for the generation of NO, which then facilitates NMDA-induced toxicity.
As potential limitations of our study, it should be mentioned that the research design here was based on the use of the ArgII-/- mice. While ArgII is deleted, there might be other active compensatory pathways, and the presence of ArgI could potentially influence the outcome. Further studies would be warranted to assess the outcome of ArgI deletion or inhibition on ischemic and excitotoxic brain damage. Further work is being pursued by various groups to better address unique mechanisms involving ArgII signaling and, consequently, roles of free radicals and NO in WT and relevant transgenic Arg mice in stroke. Furthermore, use of selective ArgII inhibitors in ischemic and excitotoxic models would be most informative to provide insights into the therapeutic implications of ArgII under such acute neurological insults/trauma.
Conclusion
By using ArgII-/- mice, this study suggests that ArgII is an important enzyme/protein that would regulate endothelial function and protect the brain from ischemic and excitotoxic damages. Therefore, targeting ArgII pharmacologically could be considered an integrative part of a multi-modal approach to fight acute brain damage excitotoxicity and ischemic brain injury.
Acknowledgments
The authors would like to thank Dr. Dan Berkowitz for providing the ArgII-/- mice, and the lab members for constructive feedback on the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (AT007429 and NS046400).
Disclosures
Authors declare that no competing interests exist.
9128
References
- Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, et al. (1993) Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature 363: 260-263.
- Szatkowski M, Attwell D (1994) Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci 17: 359-365.
- Murphy S, Gibson CL (2007) Nitric oxide, ischaemia and brain inflammation. BiochemSoc Trans 35: 1133-1137.
- Palmer RM, Ferrige AG, Moncada S (1987) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524-526.
- Schmidt K, Klatt P, Mayer B (1994) Uptake of nitric oxide synthase inhibitors by macrophage RAW 264.7 cells. Biochem J 301: 313-316.
- Matsui T, Nagafuji T, Kumanishi T, Asano T (1999) Role of nitric oxide in pathogenesis underlying ischemic cerebral damage. Cell MolNeurobiol 19: 177-189.
- Malleske DT, Rogers LK, Velluci SM, Young TL, Park MS, et al. (2006) Hyperoxia increases hepatic arginase expression and ornithine production in mice. ToxicolApplPharmacol 215: 109-117.
- Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiological Reviews 87: 315-424.
- Szabo C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6: 662-680.
- Chicoine LG, Paffett ML, Young TL, Nelin LD (2004) Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell MolPhysiol 287: 60-88.
- Meijer AJ, Lamers WH, Chamuleau RA (1990) Nitrogen metabolism and ornithine cycle function. Physiol Rev 70: 701-748.
- Nishimura H, Rosenblum WI, Nelson GH, Boynton S (1991) Agents that modify EDRF formation alter antiplatelet properties of brain arteriolar endothelium in vivo. Am J Physiol 261: 15-21.
- Steppan J, Ryoo S, Schuleri KH, Gregg C, Hasan RK, et al. (2006) Arginase modulates myocardial contractility by a nitric oxide synthase 1-dependent mechanism. ProcNatlAcadSci USA 103: 4759-4764.
- Yu H, Yoo PK, Aguirre CC, Tsoa RW, Kern RM, et al. (2003) Widespread expression of arginase I in mouse tissues. Biochemical and physiological implications. J HistochemCytochem 51: 1151-1160.
- Lim HK, Ryoo S, Benjo A, Shuleri K, Miriel V, et al. (2007) Mitochondrial arginase II constrains endothelial NOS-3 activity. Am J Physiol Heart CircPhysiol 293: H3317-H3324.
- Nelin LD, Chicoine LG, Reber KM, English BK, Young TL, et al. (2005) Cytokine-induced endothelial arginase expression is dependent on epidermal growth factor receptor. Am J Respir Cell MolBiol 33: 394-401.
- Kanda H, Kikushima M, Homma-Takeda S, Sumi D, Endo A, et al. (2008) Downregulation of arginase II and renal apoptosis by inorganic mercury: overexpression of arginase II reduces its apoptosis. Arch Toxicol 82: 67-73.
- Kitaura H, Uozumi N, Tohmi M, Yamazaki M, Sakimura K, et al. (2007) Roles of nitric oxide as a vasodilator in neurovascular coupling of mouse somatosensory cortex. Neurosci Res 59: 160-171.
- Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, et al. (2000) Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke 31: 2707-2714.
- Shah ZA, Nada SE, Doré S (2011) Hemeoxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience 180: 248-255.
- Ahmad AS, Zhuang H, Echeverria V, Doré S (2006) Stimulation of prostaglandin EP2 receptors prevents NMDA-induced excitotoxicity. J Neurotrauma 23: 1895-1903.
- Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, et al. (2006) Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res 99: 951-960.
- Moncada S, Rees DD, Schulz R, Palmer RM (1991) Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. ProcNatlAcadSci USA 88: 2166-2170.
- Toya T, Hakuno D, Shiraishi Y, Kujiraoka T, Adachi T (2014) Arginase inhibition augments nitric oxide production and facilitates left ventricular systolic function in doxorubicinâ€ÂÂÂÃÂinduced cardiomyopathy in mice. Physiol Rep 2.
- Prins HA, Houdijk APJ, van Lambalgen AA, Teerlink T, Meijer S, et al. (1998) Paradoxical changes in organ blood flow after arginase infusion in the non-stressed rat. Shock 9: 422-427.
- Steppan J, Nyhan D, Berkowitz D (2013) Development of novel arginase inhibitors for therapy of endothelial dysfunction. Front Immunol 4.
- Swamy M, Salleh MJM, Sirajudeen KNS, Yusof WRW, Chandran G (2010) Nitric oxide (NO), citrulline - no cycle enzymes, glutamine synthetase and oxidative stress in anoxia (hypobaric hypoxia) and reperfusion in rat brain. Int J Med Sci 7: 147-154.
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, et al. (1997) Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci 17: 6908-6917.
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, et al. (2001) Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. ProcNatlAcadSci USA 98: 1294-1299.
- Lynch DR, Guttmann RP (2002) Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J PharmacolExpTher 300: 717-723.
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J (1993) NMDA-dependent superoxide production and neurotoxicity. Nature 364: 535-537.
- Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. CurrOpinNeurobiol 11: 327-335.
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH (1991) Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. ProcNatlAcadSci USA 88: 6368-6371.

