Key words
|
| |
| Ofloxacin, Mucoadhesive suspension, HPMC, FTIR, Raman Spectroscopy XRD, SEM |
| |
INTRODUCTION
|
| |
| Oral controlled release (CR) dosage forms (DFs) have been developed over the past three decades due to their considerable therapeutic advantages, such as ease of administration, patient compliance and flexibility in formulation. Incorporation of the drug in a controlled release - gastro retentive dosage forms (CR-GRDF) can remain in the gastric region for several hours, which would significantly prolong the gastric residence time of drugs and improve their bioavailability, reduce drug wastage and enhance the solubility of drugs[1]. |
| |
| Several approaches are currently used to prolong gastric retention time. The goals of controlled drug delivery are to conserve and maintain effective drug concentration, eliminate night time dosage, improve compliance and decrease side effects[2]. In the present study, polymeric bioadhesive delayed gastric emptying devices have been explored. |
| |
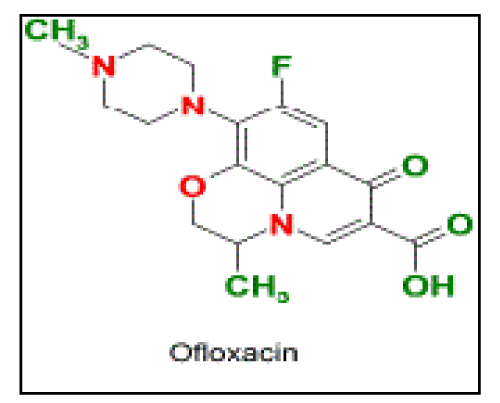
| Ofloxacin (Oflox), 9-fluro-2, 3-dihydro-3-methyl-10- (4-methyl-1-piperizinyl)-7-oxo-7H-pyrido [1,2,3-de]- 1,4-benzoxaine-6-carboxylic acid, is a fluoroquinolone antibacterial agent (Figure 1). Normal dosage regimen varies from 200 to 600 mg administered twice or thrice a day, depending on severity of infection. In severe cases, long-term therapy may also be required. Biological half-life of the drug is from 5 to 6 h. As frequent dosing is required to maintain the therapeutic plasma concentration, it was chosen as a model drug for the controlled release study[3]. |
| |
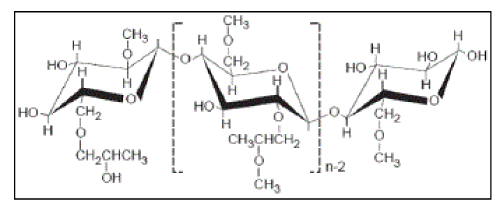
| Hydroxypropyl methylcellulose (HPMC) is propylene glycol ether of methyl-cellulose[4]. The physicochemical properties of this polymer are strongly affected by: (i) the methoxy group content; (ii) the hydroxypropoxy group content; and (iii) the molecular weight[5] (Figure 2). It is one of the most commonly used hydrophilic biodegradable polymers for developing controlled release formulations, because it works as a pH-independent gelling agent. Swelling as well as erosion of it occurs simultaneously inducing a pseudofed state, thereby reducing peristaltic contraction, which contributes to overall drug release. It is a widely accepted pharmaceutical excipient[5-9]. |
| |
| HPMC has many pharmaceutical uses, such as a drug carrier, a coating agent, a tabletting agent, and it is also used in ophthalmic solutions and in personal care products[8]. It is the most important hydrophilic carrier material used for the preparation of oral controlled drug delivery systems. One of its most important characteristics is the high swellability, which has a significant effect on the release kinetics of an incorporated drug. Upon contact with water or biological fluid, the latter diffuses into the device, resulting in polymer chain relaxation with volume expansion. Subsequently, the incorporated drug diffuses out of the system[5]. |
| |
| The HPMC may form a complex with the low solubility drug like Ofloxacin. The interaction between Oflox and HPMC can be determined by several methods such as Fourier Transform Infrared (FTIR) Spectroscopy, Raman Spectroscopy, etc. To know the different functional groups and highly polar bonds of pure Ofloxacin, HPMC, and their chemical interactions in the mucoadhesive suspension, FTIR analysis was conducted. However, their backbone structures and symmetric bonds were checked by Raman spectroscopy. Although it is known that Raman and FTIR are complementary vibrational spectroscopic techniques, there are band intensity differences between the two techniques. That is why both FTIR and Raman Spectrscopic analyses were conducted. |
| |
| The X-ray diffraction (XRD) method has become one of the most useful tools for qualitative characterization of crystalline compounds both in formulation and in pure form of the drug[10]. It is known that increased dissolution rate and delayed release of drug from dosage forms occur with increase in crystallinity[11,12]. XRD study is important because any change in the morphology of polymers, or in the crystalline state of active ingredients in the final product, resulting from the manufacturing process, can influence a drug's bioavailability[13]. |
| |
| The particle size distribution (PSD)[14] and aspect ratio (AR)[15,16] of particles in the suspension are obtained from Scanning electron microscopic (SEM) analysis. The PSD and AR distribution and degree of dispersion in the suspension give insights even into the stability relating to the modification of mechanical properties, particle-matrix interaction, polymer and drug crystallinity and the overall structure[14,17,18] of the suspension. |
| |
| Therefore, to obtain more detailed information about chemical interaction between Ofloxacin and HPMC, FTIR and Raman analyses were carried out[19,20]. Moreover, considering the bioavailability, stability and degree of dispersion of the particles present in the formulation, XRD and SEM analyses were conducted [10,13,14,17,18]. |
| |
MATERIALS AND METHODS
|
| |
|
Materials
|
| |
| The following materials were used for the study: Ofloxacin was obtained from Dr. Reddy’s Lab, Hyderabad, India, as a gift sample. Hydroxypropyl methylcellulose (HPMC E15 LV Premium) was supplied by Loba Chemie Pvt. Ltd., India. It was having methoxy group (23.8%) and hydroxypropoxy group (8.3%). Pluronic F 68 and Soya lecithin were purchased from Himedia Laboratories Pvt. Ltd., India. Glycerol, Methyl praraben sodium, Propyl paraben sodium, Sorbitol solution I.P. and Sucrose were supplied by Cosmo Chem. Laboratory, Pune, India. Ultra pure water was obtained from a Millipore Milli-Q UV water filtration system. |
| |
|
Methods
|
| |
|
Preparation of Formulation
|
| |
| · Preparation of Bulk A |
| |
| In a beaker 6 ml water was taken and it was heated up to 80° C. Sucrose (10 gm) was added to that water with continuous stirring. The temperature was monitored in such a way so that it should not fall below 70° C, till the sucrose was completely dissolved. The prepared syrup was cooled properly at room temperature and kept overnight. Syrup was filtered using 120 mesh nylon cloth. |
| |
| · Preparation of Bulk B |
| |
| Five millilitre of Ultra pure water was taken in a beaker to which 1.8 ml of sorbitol solution and 0.2 ml glycerin were added. The mixture was stirred properly. Pluronic F 68 (5%), soya lecithin (1%) and HPMC (5%) in w/w of drug were added to this solution with continuous stirring. |
| |
| · Preparation of Mucoadhesive Suspension and Ultrasonication |
| |
| Five millilitre of water was taken in another beaker to which 250 mg of Oflox was added. To the drug suspension, the bulk B and bulk A were added with continuous stirring. Methyl paraben sodium (0.015%w/v) and Propyl paraben sodium (0.08%w/v) were added as preservatives. The volume was made up to 25 ml by Ultra pure water. The pH was adjusted to 5.5. Homogenization was carried out for at least 20 min by ULTRASONIC HOMOZENIZER LABSONICR M (SARTORIUS), having operating frequency 30 KHZ and line voltage 230 V/50 HZ, using the probe made up of Titanium of diameter 7 mm and length 80 mm. The setting knob “cycle” was adjusted to 0.8, indicating sound was emitted for 0.8 s and paused for 0.2 s. In this manner, we could expose our sample with 100% amplitude, while reducing the heating effect to 80%. This LABSONICRM generates longitudinal mechanical vibrations with a frequency of 30,000 oscillations / s (30 KHZ). The probes bolted to the sound transducer were made of high-strength Titanium alloys, built as λ /2 oscillators. It amplified the vertical oscillation, and transferred the ultrasonic energy via its front surface with extremely high power density into the sample that was to be subjected to ultrasonic waves. In our study, stress applied was sound wave and in addition, mild rise in temperature of the sample occurred during ultrasonication which helped in the homogenization of the suspension. Some portion of the homogenized suspension was kept for Raman Spectroscopic analysis and SEM study. The remaining portion of the suspension was sprayed on to an aluminum slip with the aid of an atomizer. The fine droplets were dried overnight at room temperature and the solid samples were then collected and powdered. The sample was then divided into two parts –one part was for FTIR analysis, and the other part was used for XRD study. |
| |
|
Fourier Transform Infrared Spectroscopy
|
| |
| FTIR analysis was performed by FTIR Spectrophotometer interfaced with infrared (IR) microscope operated in reflectance mode. The microscope was equipped with a video camera, a liquid Nitrogen-cooled Mercury Cadmium Telluride (MCT) detector and a computer controlled translation stage, programmable in the x and y directions. Solid powder samples were oven dried at around 30°C, finely crushed, mixed with potassium bromide (1:100 ratio by weight) and pressed at 15000 psig (using a Carver Laboratory Press, Model C, Fred S. carver Inc., WIS 53051) to form disc. The detector was purged carefully using clean dry nitrogen gas to increase the signal level and reduce moisture. The spectra were collected in the 400 cm-1 to 4000 cm-1 region with 8 cm-1 resolution, 60 scans and beam spot size of 10 μm-100 μm[21-23]. The FTIR imaging in the present investigation was carried out using a Perkin Elmer Spectrum RX. |
| |
|
Raman Spectroscopic Analysis
|
| |
| The Raman system R-3000 instrument (Raman systems INC.USA), a low resolution portable Raman Spectrometer using a 785 nm solid state diode laser, was adjusted to deliver 250 mw to the sample having spectral resolution 10 cm-1 and 12 v dc/5A power supplies and USB connectivity. The solid powder samples i.e., both pure drug and polymers were enclosed in plastic poly bags and tested directly. For our study the fibre optic sampling probe was directly dipped into the formulation (prepared as per the above mentioned procedure) to collect the spectra at room temperature. The interference of the outside light was also prohibited to prevent photon shot noise. The spectra were collected over the wave number range from 140 to 2400 cm-1. |
| |
|
X-Ray Diffractometry
|
| |
| XRD measurements were obtained using the Philips X’Pert on powder diffraction system (Philips Analytical, The Netherlands) equipped with a vertical goniometer in the Bragg-Brentano focusing geometry. The X-ray generator was operated at 40 kV and 50 mA, using the CuKα line at 1.54056 Å as the radiation source. The powdered specimen was packed and prepared in a specimen holder made of glass. In setting up the specimen and apparatus, coplanarity of the specimen surface with the specimen holder surface and the setting of the specimen holder at the position of symmetric reflection geometry were assured. The powders were passed through a 100 mesh sieve and were placed into the sample holder by the side drift technique[24]. In order to prepare a sample for analysis, a glass slide was clipped up to the top face of the sample holder so as to form a wall. Each powder was filled into the holder and tapped gently. Each sample was scanned from 10° to 70° (2θ) and in stage sizes of 0.020; count time of 2.00 s, using an automatic divergence slit assembly and a proportional detector. The samples were scanned at 25° C. Relative intensities were read from the strip charts and corrected to fix slit values. |
| |
|
Scanning Electron Microscopy
|
| |
| In order to examine the particle surface morphology and shape, SEM was used. The mucoadhesive suspension (as mentioned above) was sprayed on to an aluminum slip with the aid of an atomizer. The fine droplets were dried overnight and it was used for SEM analysis [25]. The samples were given a conductive coating (using Pt, of about 600 A0 thick), using sputter ion coater and examined with SEM (JEOL JSM-6480LV) equipped with a backscattered electron detector for imaging and EDXA for elemental analysis. In this method, a focused electron beam is scanned over the sample in parallel lines. The electrons interact with the sample, producing an array of secondary effects, such as back-scattering, that can be detected and converted into an image. The image can then be digitalized and presented to an image analyzer, which uses complex algorithms to identify individual particles and to record detailed information about their morphology. Then particle size can be determined with a programme such as Image Tool or annotate either automatically or manually. Here, manual determination is preferred, because sometimes the particle boundaries are indistinct, and the software may interpret them incorrectly. The PSDs reflect the statistical result from all sections for each sample. As these are rod like particles, the aspect ratios of rod-like particles are evaluated by comparing the particle size distribution data derived from SEM analysis following the techniques described by Jennings and Parslow[17]. Length/width ratios are satisfactorily determined by the aspect ratio value. |
| |
RESULTS
|
| |
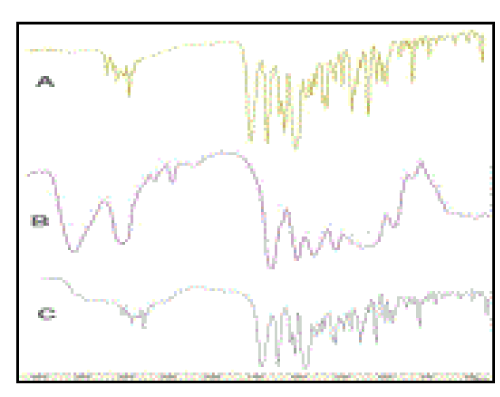
| FTIR Analysis In FTIR spectra of Ofloxacin, one prominent characteristic peak was found between 3050 and 3000 cm-1, which was assigned to stretching vibration of OH group and intramolecular hydrogen bonding (Figure 3A). This band also suggested the NH stretching vibration of the imino-moiety of piperazinyl groups which was less prominent due to intense OH stretching vibration. The peak at 2700 cm-1 was assigned to υCH3 of methyl group. The band at 1750-1700 cm-1 represented the acidic carbonyl C=O stretching i.e., υC=O[26]. The peak at 1650 to 1600 cm -1 was assigned to υN-H bending vibration of quinolones. The 1550 to 1500 cm-1 represented the υCH2 of the aromatic ring. The band at 1450-1400 cm-1 was assigned to the stretching vibration of CH2 confirming the presence of methylene group in benzoxazine ring. The peak at 1400-1350 cm-1 represented the bending vibration of hydroxyl group. The band at 1250 to 1200 cm-1 suggested the stretching vibration of oxo group. In addition, a strong absorption peak between 1050 and 1000 cm-1 was assigned to C-F group. The band at 900-800 cm-1 represented the out of plane bending vibration of double bonded ‘enes’ or =CH groups (Table 1a)[21,22,27,28]. |
| |
| Assignments of FTIR frequencies of HPMC were achieved by comparing the band positions and intensities observed in FTIR spectra with wave numbers and intensities. The peak at 3500 to 3400 cm-1 was due to OH vibrational stretching (Figure 3B)[21,22]. The symmetric stretching mode of υsMe and υshydroxypropyl groups was found in the range 2900 cm-1 in which all the CH bonds extend and contract in phase[22]. The peak at 2550-2500 cm-1 was assigned to OH stretching vibration, i.e., υO-H and intramolecular hydrogen bonding[21,22]. The band between 1650 and 1600 cm-1 indicated the presence of stretching vibration of υC-O for six membered cyclic rings. Two bending vibrations might occur within a methyl group. The first of these, the symmetric bending vibration of δsMe involved the in-phase bending of the C-H bonds. The second, the asymmetric bending mode of δasMe was due to outof- phase bending of the C-H bonds. While the asymmetric bending vibrations of the methoxy group normally appeared in the region 1500-1450 cm-1, the symmetric vibrations were mostly displayed in the range 1400-1350 cm-1 [29,30]. The band between 1400 and 1350 cm-1 suggested υC-O-C of cyclic anhydrides. The peak at 1300-1250 cm-1 was due to υC-O-C cyclic epoxide. The band at 1100-1000 cm-1 was for stretching vibration of ethereal C-O-C groups. The peak at 1000-950 cm-1 was due to υas of pyranose[31]. The rocking mode of CH2 was found in the range of 850-800 cm-1 [29] (Table 1b). The computed frequencies of HPMC are in a good agreement with experimental frequencies for both carbohydrate region as well as OH and CH region. |
| |
| In the FTIR spectra of the mucoadhesive suspension, the peak from 3100 to 3000 cm-1 was assigned to polymeric υO-H and hydrogen bonding, the band between 3000 and 2600 cm-1 represented the stretching vibration of υO-H i.e., strong intermolecular hydrogen bonding (Figure 3C). The band from 1650 to 1600 cm-1 was assigned to υC=O i.e., carbonyl stretching vibration. A prominent peak at 1500-1450 cm-1(w) was for υC-O / δO-H. The band from 1400-1350 cm-1 was assigned to δC-O-C representing esters and symmetric bending of methoxy groups. The peak between 1100 and 1000 cm-1 represented υC-F groups[21,22]. The band at 1000-950 cm-1 was assigned to υas of pyranose ring of HPMC[31] (Table 1c). |
| |
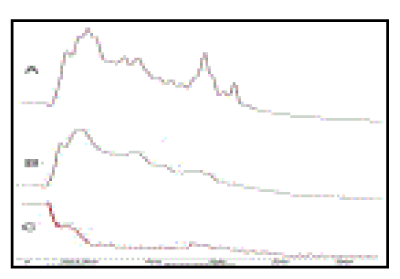
| Raman spectroscopy By Raman spectroscopy of Ofloxacin, the prominent Raman shifts were observed at 518.4, 797.5, 1419.8 and 1649.6 cm-1 (Figure 4A). The Raman shift at 518.4 cm-1 represented the bending vibration of aliphatic carbon atom, C-N stretching vibration of piperazinyl group and O-H torsional vibration of carboxylic acid. The band at 797.5 cm-1 suggested the symmetric stretching vibration of C-F group[32]. The peak at 1419.8 cm-1 was due to symmetric stretching vibration of O-C-O group of carboxylic acid and methylene deformation mode of the piperazinyl group. A band at 1649.6 cm-1 was due to symmetric stretching of the carbonyl group υC=O of the pyridone moiety, the stretching vibration of (C-C) aromatic ring chain. In addition, it (peak at1649.6cm-1) also indicated the N+H2 scissoring of piperzinyl group[33- 39] (Table 2a). |
| |
| In case of HPMC, the prominent Raman Shifts were found at 504.7, 908.3 and 1384.3 cm-1 (Figure 4B). The peak at 504.7 cm-1 was assigned to C-H out of plane bending vibration and C-C-O bending vibration of β D-glucose monomer of HPMC. The band at 908.3 cm-1 was due to C-C-C in-plane bending and υ(C-O-C) stretching vibration of pyranose ring. The peak at 1384.3 cm-1 was assigned to C-C stretching vibration (Table 2b) [29,30,33,37]. |
| |
| The characteristics Raman peaks of mucoadhesive suspension containing both Oflox and HPMC were observed at 338.8, 900-850, 1340.5 and 1800-1700 cm-1 (Figure 4C). The band at 338.8 cm-1 was assigned to C-C-C out of plane bending of pyranose ring[30]. The peak at 900-850 cm-1 was due to symmetric stretching vibration of C-F bond and symmetric COC stretching vibration for esters. The band at 1340.5 cm-1 respesented δCCH and δOCH bending vibration of methoxy group[29]. The peak at 1800-1700 cm-1 was assigned to C=O stretching vibration of carbonyl groups of esters[30] (Table 2c). |
| |
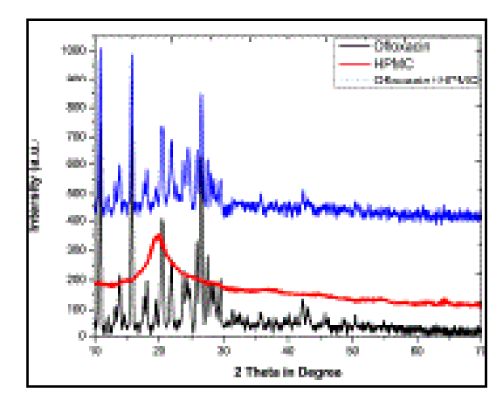
| XRD Study All the high intensity peaks (relative intensity) observed in the XRD pattern of the pure Oflox were compared with its mucoadhesive polymeric suspension (Tables 3 and 4). Both the polymeric suspension and pure Oflox were found to show similar XRD patterns (Figure 5). Identification of a structure from its powdered diffraction pattern is based upon the position of peaks and their relative intensities. Each XRD pattern is characterized by the interplanar d- spacing and the relative intensities (I/I0) of the three strongest peaks in the pattern under the Hanawalt system. The relative intensities and heights of three prominent peaks of the formulation were less than those of pure Oflox (Table 3). Moreover, complete diffraction patterns of both pure Ofloxacin and formulation can be seen in Table 4. |
| |
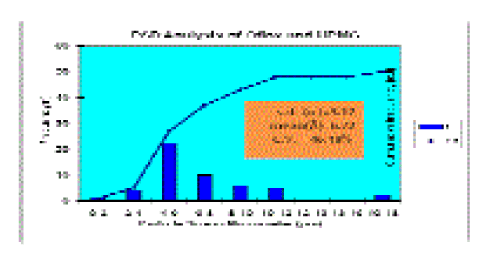
| SEM Analysis The length/width ratios of individual particles can satisfactorily determine their aspect ratios. PSD analysis of the formulation showed different ranges of length of particles along with their frequencies (Table 5, Figure 6). While within 12-16 _m range no particle was found, maximum number of particles was observed within 4-6 _m. In case of formulation, maximum aspect ratio (A.R.) frequency was found from 2 to 4 (Table 6). |
| |
| From these data (PSD and aspect ratio analysis), arithmetic means, standard deviations and coefficients of variation were calculated using standard formula[40]. The frequencies and cumulative frequencies of PSD of the formulation have been presented graphically by taking particle size ranges along X-axis, the frequencies of respective ranges on Y- axis, and the cumulative frequencies along the ? – axis (Table 5, Figure 7). The PSDs vs frequencies are plotted as histogram and PSDs vs cumulative frequencies are plotted as curves. Y- axis represents the frequencies of PSD ranges which constitutes the height of its rectangles. We get a series of rectangles each having a class interval distance as its width. The area of the histogram represents the total frequency as distributed throughout the classes. |
| |
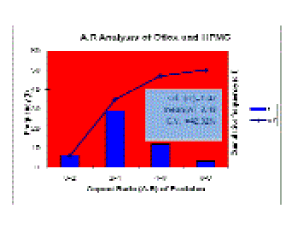
| Similarly the A.R. ranges vs frequencies and cumulative frequencies of the formulation are plotted (Table 6, Figure 8). |
| |
DISCUSSION
|
| |
| When FTIR radiation falls on a molecule, it may be absorbed, reflected or transmitted. Absorption leads to the FTIR spectrum, while reflection leads to scattering which is utilized in Raman Spectroscopy[22]. In addition, Infra red (IR) absorption of the functional groups may vary over a wide range. However, it has been found that many functional groups give characteristics IR absorption at specific narrow frequency range[21,22]. |
| |
| In case of FTIR spectra of Oflox, prominent peaks for υC-O / δO-H and υC=O indicated the presence of –CO-, -CHO and -COOH groups (Figure 3A). The presence of above groups may be confirmed by fermi resonance bands for –CHO; υC-O-C bands for esters; and absence of these two for ketones. This suggests the existence of –COOH group in Oflox (Table 1a). From FTIR spectral analysis it has been found that the HPMC shows both intramolecular and intermolecular hydrogen bonding. The presence of pyranose ring of β D-glucose monomers has been confirmed. The stretching vibration of the cyclic anhydride, methoxy and hydroxypropoxy groups along with epoxide helps in the identification of HPMC[21,22,29,30,31] (Table 1b). |
| |
| While comparing the FTIR spectra among the pure Oflox and polymer HPMC, and the mucoadhesive suspension containing both Oflox and HPMC, it is clear that the band position of C=O group has been affected by esterification and conjugation involving C=O group. Here, the stretching vibration of C=O in pure Oflox was found from 1750 to 1700 cm-1, which was lowered to 1650-1600 cm-1 in this suspension. This might be due to formation of β-ketoesters (Figure 3). The FTIR peaks assigned to υC-O and υCO- C representing esters confirm the esterification between polymeric OH group and COOH group of Oflox. The stretching vibration of C-F group remains more or less unaltered. The another probability of interaction is hydrogen bonding i.e., intermolecular hydrogen bonding due to prominent FTIR peaks between 3100 and 3000 cm-1; and 3000 and 2600 cm-1 represent polymeric O-H...O-H…O-H and strong intermolecular hydrogen bonding, respectively. The hydrogen bonded -OH stretching vibration has been found to occur over a wide range, 3100-2600 cm-1. In case of intramolecular hydrogen bonding, FTIR bands are sharp while in intermolecular hydrogen bonding bands are broad. However, it is less broad than which is required for chelation[22]. The bending vibration of O-H group indicates medium to strong bands in the region around 1450 cm-1. The peak between 1100 and 1000 cm-1 represented υC-F group of Ofloxacin [21,22].The band at 1000-950 was due to υas of pyranose ring of HPMC[31] (Table 1). |
| |
| The C=O group of drug (present in the formulation) lowers the stretching vibration of C=O frequency indicating deprotonation and probably interaction of the carboxylic C=O moiety with the polymer. However, a definite conclusion about the keto group in the bonding to the polymer can be deduced because the corresponding band found from 1650 to 1600 cm-1 is probably due to the formation of β- ketoesters[41]. From the above data it appears that the carboxylic group of Oflox undergoes the interaction with the polymer, as would be expected chemically. Thus the nitrogen atoms are not likely to be involved in binding or the interaction. Actually, the nitrogen atom of the quinolone ring, 1-ortho to fluorine, is less electron rich due to electron deficient fluoroquinolone ring. In addition, methoxy and piperazinyl groups sterically hinder the reaction. The possibility of involvement of imino moiety of the piperazinyl group is also less prominent due to intense OH stretching vibration. The bands in the region 3100-2600 cm-1 can be assigned to both asymmetric and symmetric stretching vibrations of the OH groups present in the inner and outer sphere of polymer. The shift in the characteristic bands of the FTIR spectra suggests change in their intensity leading to the appearance of several absorbance bands of the asymmetric and symmetric stretching vibrations and overtone of the deformation vibrations. This indicates the confirmation of the intermolecular hydrogen bonding. By comparing the FTIR spectra among the pure drug, HPMC polymer and the mucoadhesive suspension, the FTIR peak of pure Oflox (from 1750 to 1700 cm-1) has not been detected in the formulation, probably due to interaction with the polymer. The missing peak has been replaced by two very strong characteristic bands in the range of 1650-1600 cm-1 and at 1450 cm-1. These are assigned to υ(O-C-O) asymmetric and symmetric stretching vibrations, respectively[21,22]. The difference Δ[υ(CO2)asym-υ(CO2)sym] is a useful characteristic for determining the involvement of the carboxylic group of Oflox. The Δ value of the interaction falls in the range of 183 - 250 cm-1 indicating the deprotonation of the carboxylic acid group and interaction between drug and polymer [42] (Table 1). |
| |
| In case of Raman spectra of Oflox, the Raman band at 518.4 cm-1 is assigned to the stretching vibration of piperazinyl group and O-H torsional vibration of carboxylic acid. While the presence of carboxylic acid group is confirmed by υO-C-O at 1419.8 cm-1, the stretching vibration of υC=O groups at 1649.6 cm-1 indicates the presence of pyridone moiety (Table 2a). |
| |
| The C-H out of plane bending vibration and C-C-O bending vibration of β D-glucose monomers have been confirmed from the nondestructive Raman spectroscopic analysis of HPMC. The presence of pyranose ring is also determined by the Raman shift at 908.3 cm-1. The Raman shift for C-C stretching vibration strengthens the FTIR results for the characterization of HPMC polymeric chain[29-33]. |
| |
| By comparing the Raman spectra of pure drug with the drug incorporated in the Ofloxacin mucoadhesive suspension, the peak at 1419.8 cm-1 representing υsOC- O is not prominent. Moreover, the symmetric stretching vibration of C-O-C group and stretching vibration of C=O are prominent in our mucoadhesive formulation. From this it is clear that there is esterification reaction between the Oflox and HPMC polymer (Table 2). The results of both FTIR and Raman spectra indicate that both the spectra show prominent peaks for the stretching vibration of C-OC and C=O groups, which prove the formation of the esters between the drug and polymer. Moreover, both the intermolecular and polymeric hydrogen bondings are also prominent from the FTIR spectra of the suspension. |
| |
| Tables 3 and 4 give the data obtained for the pure Oflox, and its polymeric suspension with HPMC in terms of the lattice spacing and the relative peak intensities. Most of the characteristic peaks in the diffraction patterns are generally prominent and sharp, so measurement of the angles and d-values is accurate. |
| |
| From the XRD patterns of HPMC, it is clear that the polymer is fully crystalline in nature as there are sharp and prominent peaks (Figure 5). Table 3 confirms that the three prominent peaks of pure Oflox and its mucoadhesive suspension do not have similar d-spacing corresponding to identical 2θ values. As the d-spacing of the prominent XRD peaks of pure Oflox is changed in the polymeric composites, it may be concluded that there is interaction between Oflox and HPMC (Figure 5). However, Oflox can be easily distinguished even in the formulation. Moreover, since relative intensities of the peaks are decreased in formulation, crystallinity is also reduced in the composites as compared with pure Oflox. This decrease in relative intensities of these peaks appears to be due to change in atomic densities in that particular plane of crystal lattice. From this we may predict that there is a little bit change in the orientation of crystal lattice due to incorporation of some extra atoms into it, which may be due to hydrogen bonding and esterification. |
| |
| As we know the standard deviation measures the absolute dispersion (or variability of a distribution), a small standard deviation indicates a high degree of uniformity of the observations as well as homogeneity of a series[40]. The series, in which coefficient of variation is less, is said to be less variable, and more consistent, uniform, stable and homogeneous. From PSD study it has been found that maximum particle size of the formulation is within the pharmaceutically acceptable limit [43] (Table 4). Considering graphical analysis, the maximum particle size range for formulation containing Oflox and HPMC is between 4 and 6 _m (Figure 7). As these are rod like particles, A.R. has been calculated. From the statistical interpretation, it has been found that aspect ratios in the formulation containing Oflox and HPMC are homogeneous, consistent and stable with lesser standard deviation (Table 5, Figure 8). The mean particle size and A.R. values of the formulation (6.72 _m and 3.48, respectively) show a correlation between the particle size, particle shape and stability properties, giving confidence in the usefulness of SEM for characterizing such type of formulations [44,45]. The morphologies and mechanical properties of the formulation impart SEM sectioning and imaging, which can allow direct measurement of PSD and A.R. of particles embedded in polymeric suspension. The SEM-derived information correlated well with the mechanical properties of the present formulation. From the above SEM image analysis, it is expected that the formulation containing Oflox and HPMC is having better bioavailability and penetration capacity, as maximum particles are of A.R. values between 2 to 4 [46]. From the A.R. analysis, it can be said that the formulation is more stable because it has lesser standard deviation. Hence, it indicates that the particles in the formulation are uniformly dispersed. |
| |
CONCLUSION
|
| |
| On the basis of the above interpretation, it can be concluded that by preparing mucoadhesive suspension of Ofloxacin with HPMC following a novel method of ultrasonication, there is a very good interaction between the carboxylic group of drug and hydroxyl group of polymer. This leads to esterification and intermolecular hydrogen bonding, by virtue of which a stable mucoadhesive suspension would be produced. From the XRD data supported by FTIR analysis, it appears that the crystalline form of pure Oflox under the experimental conditions resulted in little change in crystal habit of the drug. Moreover, size of the crystals was significantly influenced by intermolecular hydrogen bonding and esterification between Oflox and HPMC. The retention of crystallinity nature of the drug in the formulation may lead to increase in stability, decrease in solubility and delay in release of the drug from polymeric suspension. This may result in controlled release action of the formulation. From the SEM image analysis, it may be concluded that the formulation containing Oflox and HPMC is having uniform dispersion of particles and stability, which may lead to better bioavailability and penetration capacity than conventional dosage forms. |
| |
| The utility of the present work may be improved if the delivery rate, biodegradation and site-specific targeting of such mucoadhesive suspension would be properly monitored and controlled. |
| |
Tables at a glance
|
 |
|
|
| Table 1 |
Table 2 |
Table 3 |
|
|
|
| Table 4 |
Table 5 |
Table 6 |
|
| |
Figures at a glance
|
 |
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
Figure 4 |
 |
 |
 |
 |
| Figure 5 |
Figure 6 |
Figure 7 |
Figure 8 |
|
| |


