Key words
|
| |
| Emulsomes, Hypertension, Liposomes, Microemulsions, Nanoparticles |
| |
INTRODUCTION
|
| |
| Novel drug delivery systems are designed to achieve a continuous delivery of drugs at predictable and reproducible kinetics over an extended period of time in the circulation. The potential advantages of this concept include minimisation of drug related side effects due to controlled therapeutic blood levels instead of oscillating blood levels, improved patient compliance due to reduced frequency of dosing and the reduction of the total dose of drug administered [1, 2]. Hence, the combination of both sustained release and control release properties in a delivery system would further enhance therapeutic efficacy. |
| |
|
Sustained and prolonged release tablets
|
| |
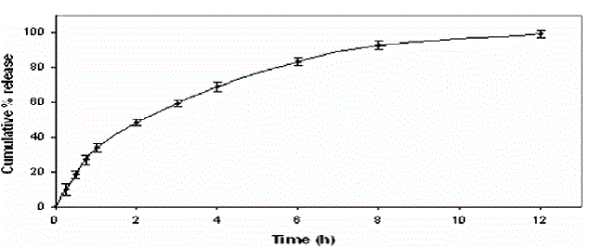
| Bioadhesive drug delivery systems are designed for prolonged retention on the mucosa to facilitate drug absorption over a prolonged period of time by interacting with mucin [3]. Anti hypertensive drug such as Losartan would be an ideal candidate for sustained drug release due to its short half-life of 1–2 hr, necessitating frequent administration of doses, as well as its severe dose dependent side effects [4]. Hydrophilic matrix tablets of LP with HPMC K15M, HPMC K100M and Na-CMC were prepared and optimized using a three-factor, three-level Box Behnken design. The formulation composition with polymer levels of HPMC-K15M, 37.40 mg, HPMCK100 M, 10 mg and Na-CMC, 24.91 mg, was found to fulfil the maximum requisite of an optimum formulation because of better regulation of % burst release and % dissolution after 1 h time interval. The optimized formulation was found to release about 99.12% drug in sustained release manner for 12 h. Study of the in vitro release profiles in 0.1 N HCl (for 2 h) and in phosphate buffer, pH 6.8 (for 10 h), of the formulations showed a burst release of 33.76% during 1 h followed by a gradual release phase for about 10 h. Fig.1 shows the complete dissolution profile of the optimized formulation. The release pattern of the optimized formulation was best fitted to both the zero order (burst release) and Korsmeyer- Peppas kinetics (sustained release phase) with R2 values of 0.9948. |
| |
| Another study for preparation of matrix tablets of losaran has also been investigated. Aim of the study was to prepare controlled release matrix tablets of Losartan potassium[5] by simplex lattice design and evaluating the relationship and influence of different content levels of HPMC, Eudragit RSPO, Eudragit RLPO and ethylcellulose, in order to achieve a zeroorder release of Losartan potassium[6,7,8]. In order to investigate the possible interactions between Losartan potassium and distinct polymers and/or diluents, FT-IR and DSC studies were carried out. DSC studies indicate that chosen excipients for the formulation were found to be compatible with the active ingredient as the melting endothermic peaks are in the range of 170- 240oC which is same as the melting point of Losartan potassium.The different polymers and/or diluents used for the preparation of matrix tablets of losartan potassium were in the ratio as shown in table 1. |
| |
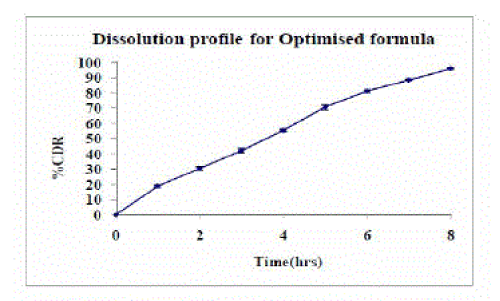
| The dissolution profile for the optimized formulation showing drug release at 1 hr was 18.87±0.72, at 4hrs was 55.50±0 and release of the drug at 8 hr was 95.92±0.57 indicating that the overall drug release from the dosage form follows zero order drug release profile as shown in figure 2. |
| |
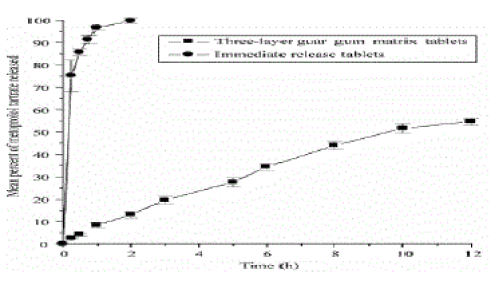
| An three-layer matrix tablet of metoprolol highly soluble drug, for oral controlled drug release, has also been reported. Metoprolol tartrate (b1-selective adrenoceptor antagonist) is widely used in the longterm treatment of hypertension and coronary heart diseases [9,10]. Metoprolol tartrate, having high solubility, is an ideal drug candidate for designing oral controlled release dosage form. It was reported that guar gum in the form of a three-layer matrix tablet could provide oral controlled release of a highly water-soluble drug such as metoprolol tartrate [11]. The three-layer guar gum matrix tablets were prepared using maximum compression force. The hardness of the tablets was 5.63 ± 0.15 kg that could contribute to the control of drug release from the hydrophilic matrix three-layer tablet formulation. When the three-layer guar gum matrix tablets were subjected to in vitro drug release studies, there was a slow drug release and kinetic evaluation of drug release showed that these oral controlled release formulations provided a first order release rate constant of 0.0696 + 0.02 h-1 which is nearer to the required release rate constant of 0.0232 h-1 calculated as per the pharmacokinetic parameters of metoprolol tartrate for twice daily administration. However, the reference immediate release tablet formulation was found to release almost all the drug immediately within 1 h (Fig. 2). Thus there existed a clear difference in the in vitro drug release characteristics of guar gum three-layer matrix tablets, which in turn, were expected to provide a slow and prolonged in vivo drug release in humans. |
| |
| The guar gum based three-layer matrix tablets of highly water-soluble metoprolol tartrate provided pseudo-steady state concentration of the drug in comparison with the immediate release tablet dosage form. The delayed Tmax; lower Cmax; decreased ka; unaltered bioavailability, and prolonged t1/2 indicated a slow and prolonged release of metoprolol tartrate from guar gum three-layer matrix tablets. |
| |
|
Nanoparticles
|
| |
| Drug encapsulated nanoparticles are solid colloidal particles that range from 10 to 1000 nm in size [12]. Based on their size and polymeric composition, they are able to target drug to specified sites in the body, and have also shown potential for sustained drug delivery [13]. Nanoparticles have also been explored for improving the formulation and efficacy of drugs with physicochemical problems such as poor solubility and stability [14]. They are being increasingly investigated for delivery of Anti hypertensive drugs to achieve sustained drug release kinetics. |
| |
| Their encapsulation into such systems may provide improved efficacy, decreased drug resistance, the reduction in dosage, a decrease in systemic toxicity and side effects, and an improvement in patient compliance. For drug nanoparticle preparations, the wet process is generally employed, and organic solvents are used in most formulations. In this study the nifedipine (NI) nanoparticles are prepared without using any organic solvent. NI nanoparticles with a mean particle size of approximately 50 nm could be prepared without organic solvent by a combination of roll mixing and high-pressure homogenization. To maintain the particle size of the nanoparticles in suspension for a long time, a method of adding gelatin powder to the NI-lipid nanoparticle suspension, dissolving the mixture by heating, and then solidifying by cooling was investigated The mean particle size of NI-lipid mixture prepared by roll mill co-grinding and subsequent high-pressure homogenization at a pass number of 40 was about 55 nm, showing that an NI-lipid nanoparticle suspension could be prepared without organic solvent.[15] |
| |
| Nano particle formulation is also used for the treatment of hypertension using coenzyme Q10. In this study Didodecyldimethylammonim bromide (DMAB) is used as a stabilizer for the preparation of nanoparticles intended for oral administration [13,14,16]. The blank nanoparticles were 100.9 ± 2.8 nm in size with polydispersity index (PDI) of 0.095 ± 0.007. With drug loading, an increase in size of particles by few nanometers was observed. No significant change was observed in the particle size, particle size distribution and zeta potential with increase in drug loading from 5% to 30%; however, a slight increase in the particle size and its distribution was observed with 50% and 75% loading. |
| |
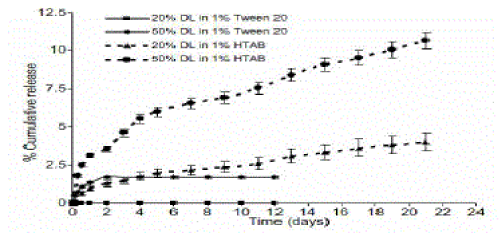
| Nanoparticles were further evaluated for in vitro drug release. In vitro release was first carried out in 1% Tween 20 solution. In 1% Tween 20 solution, the 20% CoQ10 loaded nanoparticles showed no release at all. The 50% CoQ10 loaded nanoparticles showed an initial burst effect, which continued for 48 h and resulted in around 1.7% cumulative release . However, after 48 h there was no observable release from the nanoparticles. The release studies were continued for 12 days. At the end of 12 days since no release was being observed in both the batches, the amount of CoQ10 remaining in the particles was estimated after its extraction from the nanoparticles, followed by HPLC analysis. The amount of CoQ10 remaining was 85% and 83% for 20% and 50% loaded nanoparticles, respectively. Failure of nanoparticles to release CoQ10 may be due to possible interaction or complexation between the polyethylene groups of Tween 20 with isoprene groups of CoQ10. |
| |
| The subsequent release studies were carried out in 1% hexadecyltrimethyl ammonium bromide(HTAB) solution [17], where a burst effect was observed for both 20% and 50% loaded nanoparticles. The release from 20% loaded nanoparticles was slow and about 4% of drug was released in 21 days, on the other hand 10% drug release was observed for particles with 50% loading over 21 days. This faster release observed with 50% loaded particles in comparison to 20% loaded particles can be attributed to the difference in the proportion of polymer to CoQ10 entrapped. The entrapment efficiency being very similar for both the drug loaded batches, 20% loaded nanoparticles had higher ratio of polymer to CoQ10 than the 50% loaded nanoparticles resulting in a slower release. the study was stopped at the end of 21 days The amount of CoQ10 remaining was 20% and 33% for 20% and 50% drug loaded nanoparticles, respectively: |
| |
| Thus present investigation illustrates that routinely used nutritional supplements such as CoQ10, which are generally found to be safe, can be used as first line therapeutic agents for prophylaxis and therapy by overcoming the problems associated with its delivery. CoQ10 loaded PLGA nanoparticles stabilized with DMAB administered orally have been found to be suitable for treatment of hypertension. |
| |
 |
| |
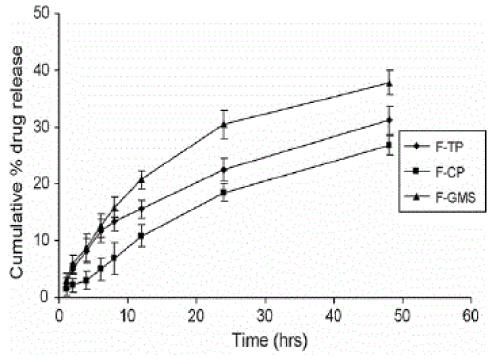
| Solid lipid nanoparticle (SLN) were prepared by homogenization followed by ultrasonication. They play role to increase the bioavaibility and for controlled release of antihypertensive drugs. Nitrendipine is an antihypertensive drug with poor oral bioavailability ranging from 10 to 20% due to the first pass metabolism. For improving the oral bioavailability, nitrendipine loaded solid lipid nanoparticles have been developed using triglyceride, monoglyceride and wax. Polaxamer 188 is used as a surfactant. To obtain stable and smaller SLN, poloxamer concentration was varied from 0.5% to 1.5%. poloxamer concentration of 1% was effective in producing smaller size SLN in case of all the three lipids (110.6–115.4 nm for F-TP, 116.4–122.3 nm for F-CP and 132.6–137.4 nm for F-GMS. The entrapment efficiency of theSLN for nitrendipine was in the order of F-TP > F-CP > F-GMS. The AUC(0–∞) of all three SLN formulations (6.65±0.24_g-h/ml with F-GMS, 6.24±0.11_g-h/ml with F-TP and 5.23±0.1_g-h/ml with FCP) were significantly (p < 0.05) higher than that of nitrendipine suspension suspension (1.69±0.13µg-h/ml). Percentage of nitrendipine released from SLN formulations up to 48 h was 31.12% with FTP, 26.7% with F-CP and 37.8% with F-GMS. Interestingly, the particle size had no influence on the in vitro release of nitrendipine as F-GMS with larger particle size (136.2 nm) showed high release (37.8%) as shown in Fig 4.The release of a drug from the SLN can be influenced by nature of the lipid matrix, surfactant concentration and production parameters (Muller et al., 2000). |
| |
| The fig 4 shows that formulation prepared from glyceryl monostearate shows maximum release (37.8%) even the F-GMS has larger particle size (136.2nm). |
| |
| Microemulsion: Microemulsions have been studied for delivery of Anti hypertensive drug such as lacidipine (LCDP), a poorly water soluble and low bioavailable drug[18]. The bioavailability studies in rabbits showed that about 3.5 times statistically significant (p < 0.05) improvement in bioavailability, after transdermal administration of microemulsion gel compared to oral suspension. Microemulsion based transdermal therapeutic system of LCDP was developed and optimized using Box–Behnken statistical design and could provide an effective treatment in the management of hypertension. IPM (isopropyl myristate) containing 10% (v/v) DMF (dimethyl Formamide) was selected as oil phase to solubilize LCDP that could further maintain large concentration gradient towards skin.The solubility of LCDP found to be 42.83 in IPM containing 10% (v/v) DMF [19]. LCDP is released and permeated well from microemulsion gel by transdermal route, as compared to the oral suspension. This difference was because of stratum corneum that could delay the permeation of LCDP from microemulsion gels in contrast, suspension administered by oral route is an immediate release dosage form. The developed microemulsion gel formulation was efficacious for the delivery of lipophilc and poorly soluble drugs such as lacidipine. The results demonstrated that the formulation was nonirritating and did not cause any erythyma upon transdermal administration. |
| |
|
Microsphere:
|
| |
| Microspheres are spherical empty particles with size varying from 50 nm to 2 mm. The microspheres are characteristically free flowing powers consisting of synthetic powder, which are biodegradable in nature ideally having a particle size less than 200 µm. Traditional microsphere drug delivery systems using a single polymer have several inherent flaws such as high initial burst, low encapsulation efficiency for highly water soluble drugs, inability to lend themselves to pulsatile or zero order release and lack of sustained release for periods suitable for periodic therapy. Composite double-walled microspheres adapted for the encapsulation of a highly watersoluble, have the ability to circumvent some of these Limitations[20]. Aim of the present study is the development of Double walled microspheres for sustained release of anti hypertensive drugs. For this study, have selected polymer: Chitosan and Eudragit E100. The inner core which is made up of polymer chitosan will contain drug; propranolol hydrochloride and outer shell which is made up of polymer Eudragit E100 contain; furosemide. Since Eudragit E100 is dissolving below pH5, will release the drug (furosemide) and attain therapeutic plasma concentration, which reduces the body fluid thus also reduces blood pressure then the inner core chitosan’s is a mucoadhesive, contains propranolol hydrochloride, adhesive to mucous layer of Stomach or GIT will provide the sustain release of the drug for a longer period (24 hr). |
| |
| Another investigation also describes preparation of microspheres shows sustained release, the morphology (Scanning Electron Microscopy), particle size distribution, total entrapment of Losartan potassium into the microparticles and their release profiles were investigated[21].The drug carrier interaction were investigated in solid state by FT-IR spectroscopy and HPLC study[22]. The Losartan loaded microspheres were prepared by solvent evaporation method using different combination of primary viz. ethyl cellulose and alginate and secondary polymers viz. different grades of acryl coats as described in the Table 1. |
| |
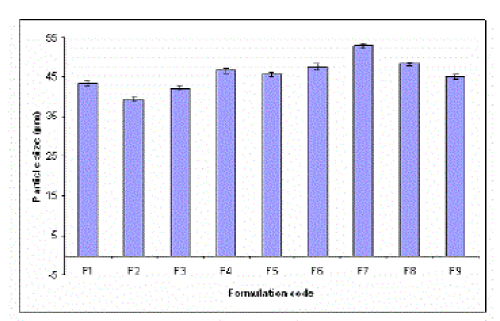
| The percentage yield of all the formulation was found to be satisfactory and drug entrapment efficiency (DEE) of all formulations were found to be more than 80 %.Using optical microscopy method[23,24,25] the mean geometric particle size of microspheres was found in a range of 40 to 50 ìm . |
| |
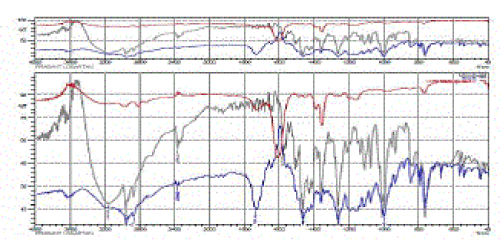
| The interaction study between the drug and polymers in different formulations were performed using FTIR spectrophotometer andHPLC. The drug shows different peaks at C-H = 3008, C=C = 1605, 1495, 1466, O-H = 3231, N=N = 1576 and Cl = 1200- 1400cm -1of benzene which confirms the purity of the drug. FT-IR spectrum of pure Losartan potassium and formulations (F3) is represented in Fig 6. |
| |
| The polymer combinations of ethyl cellulose, alginate and acrylic release retardant polymers resulted in microspheres with good yield and moderate entrapment. Results of the present study suggest that combinations of both polymers in different ratio shows sustained release microspheres. |
| |
Conclusion
|
| |
| Novel drug delivery systems will present an opportunity for formulation scientists to overcome the many challenges associated with current antihypertensive drug therapy, thereby improving the management of patients with hypertension in future. |
| |
Conflict of Interest
|
| |
| NIL |
| |
Source of Support
|
| |
| NONE |
| |
Tables at a glance
|
 |
|
| Table 1 |
Table 2 |
|
| |
Figures at a glance
|
 |
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
Figure 4 |
 |
 |
 |
| Figure 5 |
Figure 6 |
Figure 7 |
|
| |


