Keywords
Kabuki syndrome; Congenital abnormalities; Congenital constricted ears
Introducción
El síndrome de Kabuki o síndrome de Niikawa-Kuroki [1,2] (antes conocido como el síndrome de maquillaje de Kabuki), comprende una serie de anormalidades de las cuales la más característica son los rasgos faciales. La denominación de este síndrome, se debe al parecido físico de estos niños con el maquillaje de los actores del teatro tradicional japonés Kabuki, especialmente por el aspecto de la eversión del borde inferior externo de los párpados [3].
El síndrome de Kabuki (SK) es un síndrome con retraso mental congénito (leve a moderado) y con retraso del crecimiento postnatal [1]. Los rasgos faciales característicos incluyen fisuras palpebrales alargadas con eversión de los párpados inferiores, pestañas prominentes, adelgazamiento lateral de las cejas, pabellones auriculares prominentes [4], una punta nasal ancha y deprimida, lóbulos de las orejas prominentes. Otras anormalidades corporales incluyen la persistencia del botón de la yema del dedo fetal, alteraciones del esqueleto, paladar hendido, laxitud articular, anomalías dentales, infecciones recurrentes, patrones inusuales dermatoglíficos, así como anomalías cardiacas y viscerales [5].
La prevalencia del síndrome se estima de 1 por cada 32 mil en la población japonesa [1], con una proporción hombre mujer de 1.16:1, con una incidencia general de uno entre cada 32 mil y 86 mil personas. Contando actualmente con más de 300 casos descritos en la literatura internacional [2] y en México con 3 casos reportados previamente [3,6,7].
Presentación del Caso
Se trata de paciente masculino de 12 años de edad quien presenta los siguientes antecedentes de importancia: Producto obtenido por parto eutócico quien requirió reanimación neonatal, con peso de 2800 gr, 49 cm de altura, se desconoce Apgar y Silverman, antecedente de cierre de paladar blando a los 6 años de edad, antecedente de bronconeumonía y retraso del desarrollo psicomotor especialmente en el área del lenguaje. A la exploración física se valoró paciente de 25 kg de peso con 126 cm de altura (ambos con percentila 5 para población mexicana), con alargamiento de las fisuras palpebrales, inclinación antimongólica, arqueamiento de cejas, telecanto, punta nasal ancha y deprimida, almohadillas fetales, camptodactilia del 5to dedo en ambas manos (Figuras 1 y 2), microtia y apéndices preauriculares únicamente derechos; y retraso del desarrollo psicomotor con especial enfoque en el aspecto de interacción y lenguaje. Fenotípicamente sin alteraciones adicionales. Valorado por el servicio de Genética, descartando alteraciones renales y cardiacas mediante ecografía. Por las características clínicas se estableció el diagnóstico de síndrome de Kabuki con un cariotipo normal, siendo enviado al servicio de Cirugía Reconstructiva por presentar malformación de pabellón auricular por presencia de oreja constreñida II A de acuerdo a la clasificación de Tanzer (Tabla 1) para oreja constreñida y poliotia, ambas del lado derecho, la oreja izquierda sin alteraciones.

Figure 1: Fisura palpebral alargada, inclinación antimongólica, arqueamiento de cejas y presencia de punta nasal ancha y deprimida, rasgos característicos del síndrome de Kabuki.
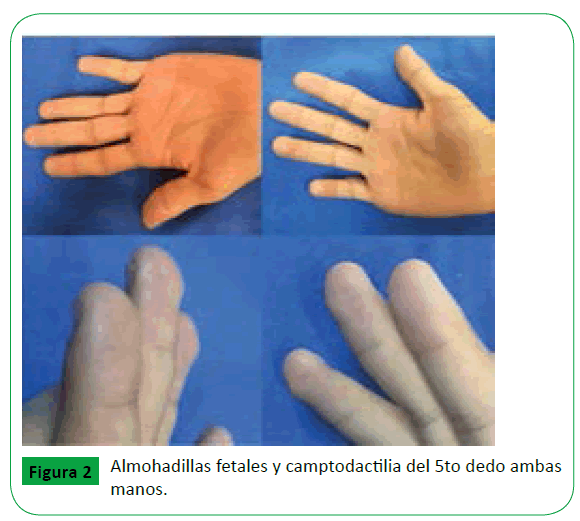
Figure 2: Almohadillas fetales y camptodactilia del 5to dedo ambas manos.
| Grupo |
Descripción |
| I |
Involucro sólo del hélix |
| II |
Involucro del hélix y la escafa |
| II A |
Sin necesidad de suplemento de piel en el margen auricular |
| II B |
Necesidad del suplemento de piel en el margen auricular |
| III |
Deformidad en copa extrema; generalmente asociada con migración incompleta, inclinación hacia delante, estenosis del conducto auditivo externo y sordera |
Tabla 1 Clasificación de Tanzer para oreja constreñida [18].
Discusión
Recientemente, se ha demostrado dependiendo de la corte estudiada, que mutaciones en el gen que codifica para la proteína mieloide/linfoide o de linaje-mixto leucemia 2 gen (MLL2) localizado en el cromosoma 12 (12q13.12) y el gen KDM6A que codifica para la desmetilasa lisina específica tipo 6A en Xp11.3, son responsables del síndrome de Kabuki [8]. Un loci alternativo se sugirió como responsable en el resto de pacientes. En la mayoría de los casos del síndrome de Kabuki (60% de los casos) se trata de un trastorno autosómico dominante con penetrancia completa, pero con algunas diferencias en los patrones de expresión. Desde su descubrimiento, se han reportado más de 100 mutaciones diferentes en el gen MLL2, principalmente mutaciones sin sentido y del marco de lectura, y en menor proporción, mal sentido y de empalme menor. Las mutaciones son esparcidas a lo largo de los 54 exones del gen sin hotspots mutacionales.
El descubrimiento de mutaciones MLL2 en la mayoría de los pacientes permite la detección en pacientes con manifestaciones fenotípicas leves. No se han informado correlaciones genotipofenotipo, dadas las funciones epigenéticas de la proteína afectada. La presencia de la mutación MLL2 sólo demostró una mayor tendencia a anomalías renales y faciales. Las diferencias fenotípicas en función del tipo de mutación aún no han sido determinadas. Sin embargo, el diagnóstico precoz es importante, ya que facilita la investigación de los problemas médicos asociados y la terapia sintomática necesaria [9].
Clínicamente los pacientes pueden presentar una variedad de anomalías que orientan al diagnóstico, las 5 características cardinales definidas por Niikawa et al [10] son; anomalías específicas del rostro, anomalías del esqueleto, anormalidades dermatoglíficas, daño mental moderado y deficiencia del crecimiento postnatal.
De las anomalías craneofaciales, la fisura palpebral larga o euriblefaron constituye las más frecuente hasta en un 95%, seguido de la eversión de los párpados inferiores laterales, las cejas arqueadas con pelo escaso en la mitad lateral exterior, malformaciones auriculares y una punta nasal deprimida. Paladar hendido u ojival y dentición anormal, incluyendo hipodoncia (especialmente faltan los incisivos), microdoncia y forma anormal de los incisivos superiores (forma de desatornillador) se observan en aproximadamente la mitad de los casos. Los hallazgos faciales menos frecuentes incluyen escleróticas azules, estrabismo, micrognatia, ptosis de los párpados y una raíz nasal amplia.
Las anomalías de las extremidades con persistencia de las almohadillas fetales o pads en los dedos son una característica clave para el diagnóstico, presentes en la mayoría de los pacientes (82%) [11], también se han descrito braquidactilia, sindactilia [12], camptodactilia y clinodactilia.
Las cardiopatías congénitas son las más frecuentes (32%) [11] con incidencia de defectos del septum ventricular, coartación de la aorta, defecto del septum atrial [10] y la válvula aórtica bicúspide.
El espectro de las enfermedades hepáticas asociadas con el síndrome de Kabuki puede involucrar una colestasis neonatal transitoria [13]. En largas series de pacientes, se ha reportado la incidencia de problemas hepáticos del 2 al 21% [14].
Las anomalías neurológicas incluyen retraso mental casi universalmente (93%), que van de leve a moderado en la mayoría, y severo en la minoría de los pacientes. Ocasionalmente presentan craneosinostosis y deformación vertebral.
En el crecimiento, aunque el peso al nacer y la longitud son generalmente normales, el retraso a menudo comienza temprano durante el primer año de vida, con una estatura final corta en la mayoría [11]. Se cree que niveles anormales en la hormona de crecimiento son el mecanismo patogénico del retraso en el crecimiento [15].
En el aspecto inmunológico, la función de las células T suele conservarse normal pero hay alteración de las células B con afección de la inmunidad humoral. En el 80% de los pacientes hay disminución de los niveles de IgA, niveles bajos de IgG en 40% y valores de IgM normales o levemente afectados [6].
Otras malformaciones renales, malrotación intestinal, anomalías anorectales, convulsiones, anomalías endocrinológicas [2] y dermatológicas como la aplasia cutis se han descrito [16].
Son muy pocos los tumores malignos reportados en asociación con el síndrome de Kabuki. Estos incluyen: un caso de virus de Epstein-Barr positivo con linfoma de Burkitt; un caso de leucemia linfocítica aguda; y un caso de neuroblastoma. Actualmente no hay conocimiento sobre si se incrementa el riesgo de cáncer [17].
Las anomalías craneofaciales no poseen un manejo quirúrgico establecido ya que el tratamiento va de acuerdo a la severidad de la malformación. En el caso expuesto, el paciente cuenta con el antecedente de cierre del paladar blando y nueva valoración por su edad para realizar reconstrucción auricular. Al paciente se le realizó moldeamiento del pabellón auricular derecho (Figura 3) con utilización de cartílago autólogo costal así como excéresis de poliotia. La excéresis consistió en la disección del componente cartilaginoso de la piel, la resección del cartílago en su totalidad (esto incluye la resección del cartílago en plano profundo), disección cuidadosa evitando lesión de ramas del nervio facial y finalmente remodelación de la piel. Se realizó un seguimiento inicial del paciente a 6 meses y posterior de 4 años en la que se observa la mejoría en el contorno del hélix posterior a la remodelación (Figura 4).
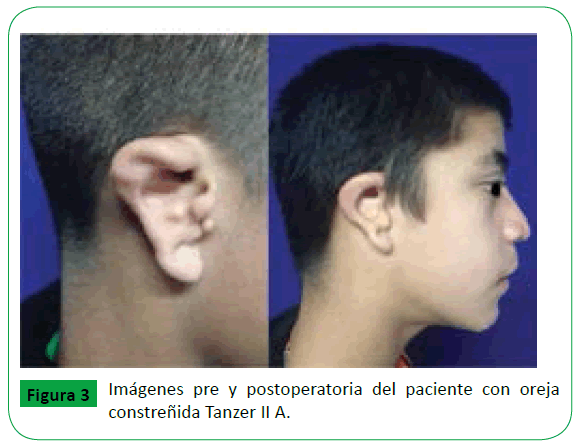
Figure 3: Imágenes pre y postoperatoria del paciente con oreja constreñida Tanzer II A.
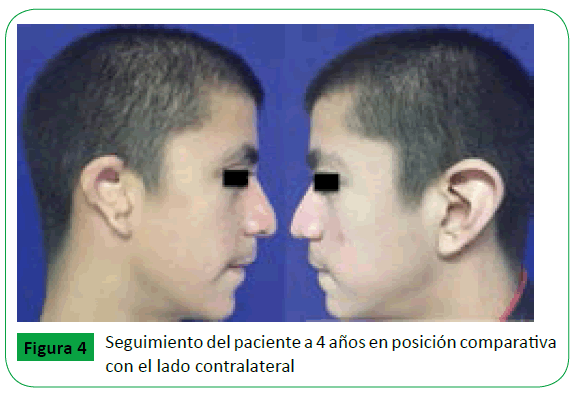
Figure 4: Seguimiento del paciente a 4 años en posición comparativa con el lado contralateral
La reparación auricular va en función al grado del defecto que presente el paciente, en el caso antes descrito presentaba involucro del hélix, escafa y antihélix pero con piel suficiente para cubrir el defecto una vez realizada la corrección quirúrgica clasificándolo como 2A de acuerdo a la clasificación de Tanzer [18].
Las opciones quirúrgicas para el tipo de microtia presentada pueden incluir la liberación del cartílago del hélix plegado, realizar incisiones radiales al cartílago o bien ambos procedimientos con reforzamiento del cartílago obtenido de región auricular posterior [19].
En el caso del paciente se optó por la toma de cartílago costal por tener mayor sitio donador y menor morbilidad del resto del cartílago auricular, el cual a pesar de estar presente en los 2 tercios inferiores no le confiere la forma habitual de una oreja sin patología.
A pesar de que la microtia no es uno de los datos característicos del síndrome de Kabuki, existen reportes que sugieren agregar esta característica fenotípica a las ya descritas, sin dejar de tomar en cuenta los diagnósticos diferenciales como el síndrome de Treacher Collins y el síndrome de Goldenhar [20].
Aunque la poliotia se ha asociado con malformaciones renales, de forma aislada no se encuentra indicado la realización de ultrasonografía renal de forma rutinaria, sin embargo dadas las alteraciones craneofaciales y la integración de un síndrome genético es importante descartar otro tipo de alteraciones a nivel renal. Otra de las asociaciones con mayor importancia es la hipoacusia, presente en el 15-30% de los que presentan poliotia. Dicha característica se encuentra presente por la deficiencia del conducto auditivo externo.
Además de estar descrito en el síndrome de Kabuki como rasgos poco frecuentes, los apéndices preauriculares pueden asociarse a las siguientes entidades; síndrome de Townes-Brocks, síndrome braquio-oto-renal, síndrome de Treacher Collins, síndrome de Miller, síndrome de Wolf, síndrome de Charge, síndrome de Di George y síndrome de Goldenhar [21,22].
También es importante tener en mente los diagnósticos diferenciales genéticos en los que se pueden hallar un anillo del cromosoma X asociado a un fenotipo Turner inusualmente severo con parecido facial al síndrome de Kabuki, presentando también fisuras palpebrales largas. Algunas de las características del síndrome de Noonan incluyendo cuello ancho, la implantación baja del cabello, orejas de implantación baja, ptosis palpebral y la teletelia también se han reportado en el síndrome de Kabuki. También tiene cierto parecido facial con síndrome Hardikar, un síndrome poco frecuente, con fisuras palpebrales largas, con enfermedad hepática obstructiva y retinopatía [11].
La historia natural a largo plazo del síndrome de Kabuki no se conoce, el pronóstico para la supervivencia en la edad adulta es bueno, sobre todo si los defectos congénitos del corazón e infecciones se traten adecuadamente en la infancia.[2] Los primeros casos descritos actualmente se encuentran en la cuarta década de la vida [6].
A pesar de los avances en el ámbito genético el diagnóstico continúa siendo clínico, orientado en las cuestiones dismórficas del paciente [6,17]. Existen casos en los cuales el paciente no tiene todos los aspectos cardinales, explicando la falta de reportes previos sobre el manejo de los pacientes, debido al gran espectro fenotípico presente [23]. Puede ser difícil de diagnosticar en el período neonatal ya que el fenotipo generalmente se hace evidente en el primer año de vida. Hay un número cada vez mayor de anormalidades fenotípicas por lo que los datos cardinales no son necesarios para establecer el diagnóstico [24]. En el caso presentado se descartaron anomalías cardiacas y renales mediante una ecografía y actualmente sólo se encuentra en seguimiento por la remodelación del pabellón auricular y por terapia de lenguaje, evolucionando de forma satisfactoria.
Existen pocos reportes sobre el síndrome de Kabuki con datos fenotípicos como la microtia y como la poliotia aunque hasta el momento en ninguno de ellos se describe la combinación de ambos. Al momento de la realización del trabajo, es el primer reporte con ambas características simultáneas por lo que se sugerimos se debe considerar como parte de los diagnósticos etiológicos que involucran estas malformaciones.
Conclusión
El manejo del paciente con síndrome de Kabuki debe ser multidisciplinario. La importancia de su conocimiento y difusión permite el diagnóstico y la búsqueda intencionada de las anomalías más frecuentes a fin de ofrecer el mejor tratamiento para una óptima calidad de vida de los pacientes. En el presente reporte se agrega la coexistencia de microtia con poliotia unilateral a las características fenotípicas ya descritas con anterioridad, por lo que sugerimos considerar el síndrome de Kabuki como posible diagnóstico ante estos rasgos craneofaciales.
Conflictos de Intere?s
Los autores declaran no tener conflictos de intere?s con la publicacio?n de este artículo.
20427
References
- Maas NM, Van de Putte T, Melotte C, Francis A, Schrander-Stumpel CT, et al. (2007) The C20orf133 gene is disrupted in a patient with Kabuki syndrome. J Med Genet 44:562-569.
- Ramachandran M, Kay RM, Skaggs DL (2007) Treatment of hip dislocation in Kabuki syndrome. J PediatrOrthop 27:37-40.
- Trigueros B, Páez H (2010)Síndrome de Kabuki; Informede uncaso en México. Rev MexOftalmol 84:176-180.
- Hoffman JD, Zhang Y, Greshock J, Ciprero KL, Emanuel BS, et al. (2005) Array based CGH and FISH fail to confirm duplication of 8p22-p23.1 in association with Kabuki syndrome. J Med Genet 42:49-53.
- Pottinger CC, Stiff RE, Holroyd JM, Davies SJ (2009) Further evidence of dominant inheritance of Kabuki syndrome. ClinDysmorphol 18:215-217.
- Aviña J, Pérez N (2006)Síndrome de Kabuki. Informe de uncaso. ActaPediatrMex 27:349-351.
- Valdez C, García S, Valdez B, García O (2013)Síndrome de Kabuki o maquillajedel teatrojaponés. Análisis de uncaso. RevistaSalud Quintana Roo 6:21-24.
- Lederer D, Shears D, Benoit V, Verellien-Dumoullin C, Maystadt I (2014) A three generation X-linked family with Kabuki syndrome phenotype and a frameshit mutation in KDM6A. Am J Hum Genet 164A:1289-1292.
- Brackman F, Krumbholz M, Langer T, Rascher W, Holter W, et al. (2013) Novel MLL2 mutation in Kabuki syndrome with hypogammaglobulinemia and severe chronic thrombopenia. J PediatrHematolOncol 35:314-316.
- Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T (1981) Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 99:565-569.
- Wessels MW, Brooks AS, Hoogeboom J, Niermaijer MF, Willems PJ (2002) Kabuki syndrome: A review study of three hundred patients. ClinDysmorphol 11:95-102.
- Elliot AM, Reed MH, Evans JA, Cross HG, Chudley AE (2004)Cenani-Lenz syndactyly in a patient with features of Kabuki syndrome. ClinDysmorphol 13:143-150.
- Isidor B, Rio M, Mourier O, Habes D, Amiel J, et al. (2007) Kabuki syndrome and Neonatal cholestasis: Report of a New case and review of the literature. J PediatrGastroenterolNutr45:261-264.
- Suskind DL, Finn L, Wahbeh G, Christie D, Horslen S (2006)Achild with kabuki syndrome and primary sclerosing cholangitis successfully treated with ursodiol and cholestryamine. J PediatrGastroenterolNutr 43:542-544.
- Gabrielli O, Bruni S, Bruschi B, Carloni I, Coppa GV (2002) Kabuki syndrome and growth hormone deficiency: Description of a case treated by long-term hormone replacement. ClinDysmorpho 11:71-72.
- Canhan NL (2006) Cutis aplasia as a feature of Kabuki syndrome. ClinDysmorphol 15:179-180.
- Shahdadpuri R, O’Meara A, O’Sullivan M, Reardon W (2008) Low-grade fibromyxoid sarcoma: Yet another malignancy associated with Kabuki syndrome. ClinDysmorphol 17:199-202.
- Tanzer RC (1975)The constricted (cup and lop) ear. PlastReconstrSurg 55:406-415.
- Nagata S (2002) Alternative surgical methods of treatment for the constricted ear. ClinPlastSurg 29:301-315.
- Chin-To F, Wang M, Young EC, Hogan CA, Tallents RH, et al. (2001)Microtia associated with the Kabuki (Niikawa-Kuroki) syndrome. Otolaryngol Head Neck Surg 125:557-558.
- Novoa A, Garrido J (2006)Niños con apéndices y fositaspreauriculares, enfoqueprácticopara el pediatra. ArchArgentPediatr104:185-188.
- Sánchez A, Ricomà V, Sampériz C (2016)Apéndicespreauriculare. ORL Aragon 19: 27-28.
- Sakurai H, Nozaki M, Takeuchi M, Soejima K, Kajimoto M, et al. Periorbitalcorrection in Kabuki syndrome. PlastReconstrSurg 111:1461-1464.
- McGaughran J, Aftimos S, Jefferies C, Winship I (2001) Clinical phenotypes of nine cases of Kabuki syndrome from New Zealand. ClinDysmorphol 10:257-262.

