Keywords
Myeloproliferative disorders; Chronic myeloproliferative síndromes; Polycythemia Vera; Primary mielofibrosis; Essential thrombocytosis; Januskinase2
Introduction
Los llamados Síndromes Mieloproliferativos Crónicos (SMC): policitemia vera (PV), trombocitemia esencial (TE), mielofibrosis primaria (MFP) y leucemia mieloide crónica (LMC) [1], se han convertido en un paradigma diagnóstico y un reto terapéutico para la salud de los pacientes; debido al poco conocimiento médico, a la poca magnitud de su prevalencia y a su importancia como factor determinante de la calidad de vida de las personas [2].
El aumento de la expectativa de vida en los últimos años, los avances técnicos, diagnósticos y terapéuticos de la medicina, así como el progresivo envejecimiento de la población son los principales responsables del aumento de la incidencia de las enfermedades crónicas invalidantes y oncológicas en los últimos años [3]. Todas estas enfermedades abocan al enfermo hacia una situación terminal irreversible y, finalmente, a la muerte [4]. Se calcula que cada año se diagnostican 7 millones casos nuevos de cáncer en el mundo de los cuales 5 millones morirán a causa del mismo, representando el 20% de todas las muertes [3]. El cáncer es la segunda causa de muerte en nuestro país (Colombia), situándose la incidencia en cifras de 250/100.000 habitantes [5].
Atendiendo a esta problemática, el aumento de la prevalencia de los síndromes mieloproliferativos en los últimos años y teniendo en cuenta que es un motivo para desarrollar cáncer en todas las líneas hematopoyéticas [6]; se hace necesario el estudio y abordaje de la población adulta con policitemia vera (PV), trombocitemia esencial (TE), Mielofibrosis primaria (MFP). Que son las patologías demostrables con mutación JAK2 para su diagnóstico: fuerte (PV), moderada o débilmente (TE, MFP) [7,8]. Se estima que cada uno tiene una tasa de 0.5 a 2.5 por 100,000 personas por año (8). Excluyendo de la presente revisión a la Leucemia mieloide crónica (LMC) debido a que su diagnóstico se hace con la aparición del cromosoma de Philadelfia [9].
La OMS incluye estos síndromes, entre las neoplasias mieloproliferativas por su origen clonal e integrando los nuevos hallazgos moleculares para su diagnóstico [10]. Con el descubrimiento de las mutaciones del gen JAK2, un nuevo paradigma en el tratamiento va orientado; y, además, en la adquisición de nuevos conocimientos etiopatogénicos que aseguran un diagnóstico certero en estas patologías.
Son numerosos los estudios que se han hecho con respecto a la frecuencia de la mutación JAK2 y los síndromes mieloproliferativos crónicos a nivel mundial. Dentro de la revisión literaria realizada, cabe destacar el estudio realizado por Gorbenko, A y col en el año 2019 [11], y el estudio realizo por Li, Z. L y col en el año 2018 [12], quienes se fundamentaron en la elaboración de estrategias que ayudaran al afrontamiento de las patologías mieloproliferativas, por medio de una revisión que se basó esencialmente, en cómo se puede diagnosticar la frecuencia de esta enfermedades gracias a la mutación de genes, en especial el JAK2 en policitemia Vera, no dejando a un lado a la trombocitopenia esencial y a la mielofibrosis primaria.
Los pacientes con neoplasias mieloproliferativas (NMP) experimentan una reducción de la supervivencia y la calidad de vida debido a eventos cardiovasculares trombóticos y/o transformación a leucemia mieloide aguda (LMA) [7,8]. Además, la mutación JAK2 que alberga una gran parte de los pacientes con NMP, se ha asociado con un mayor riesgo de linfoma [12].
Independientemente, el descubrimiento de JAK2 ha reforzado la contribución patogénica de la señalización JAK-STAT en síndromes mieloproliferativos crónicos (SMC) e identifica a JAK2 como un objetivo farmacológico válido. Realizamos una búsqueda sistemática del alcance de la literatura publicada, para investigar las características clínicas, diagnóstico y tratamiento de los pacientes con SMC JAK2 positivos.
Metodología
Se realizó una búsqueda sistemática de la literatura en pubmed, Science Direct, Nature y Google académico a través de las palabras claves del MeSH: “Myeloproliferative disorders”, “Chronic myeloproliferative síndromes”, “Polycythemia Vera”, “Primary mielofibrosis”, “Essential thrombocytosis”, “januskinase2”. La búsqueda se basó en ensayos clínicos, estudios descriptivos, revisiones sistemáticas, tesis y revisiones de la literatura. Se incluyeron artículos desde el año 2000 hasta el año 2020. Se excluyó de la presente revisión a la Leucemia mieloide crónica (LMC), debido a que su diagnóstico se hace con la aparición del cromosoma de Philadelfia y no mediante mutación JAK2. Después de la lectura crítica de los artículos seleccionados y del diseño de mapas conceptuales se procedió a la elaboración del documento.
Resultados y Discusión
En 1975, el grupo de estudio PV publicó un conjunto de "criterios de diagnóstico" que se utilizaron principalmente para garantizar la exclusión, a los protocolos de tratamiento, de pacientes con policitemia secundaria o aparente. Estos criterios requerían la demostración de masa de glóbulos rojos (RCM) incrementada mediante la medición del volumen sanguíneo utilizando eritrocitos marcados, así como la demostración de la saturación de oxígeno [13,14]. En 2001, la OMS publicó un criterio diagnóstico relacionado para PV y MFP, que reconocieron el valor de la histología de la médula ósea y los parámetros biológicos "específicos de neoplasia mieloproliferativa" [14].
En 2005, se describió una nueva mutación de Janusquinasa 2 (JAK2) también conocida como (JAK2 V617F) en asociación con PV, TE y MFP (7). Además, la mutación se encuentra en una pequeña proporción de pacientes con otras neoplasias mieloides, incluidas las neoplasias mieloproliferativas no clásicas y los Síndromes mielodisplásicos [15]. Otras mutaciones de JAK2 (es decir, mutaciones del exón 12 de JAK2) se describieron posteriormente en la mayoría de los pacientes con JAK2 V617F positivo y PV negativa o "eritrocitosis idiopática", lo que aumentaba la posibilidad de que una mutación JAK2 sea esencial para el fenotipo PV [11,12].
Polycythemia Vera Study Group (PVSG) estableció los criterios diagnósticos para TE [16]; y en el 2001 la OMS propuso nuevos criterios que fueron actualizados en el 2008. Los nuevos criterios de la OMS 2008 divide a las neoplasias mieloproliferativas crónicas clásicas BCR-ABL positivas y negativos, y en JAK2 positivas y negativas [17]. En la Figura 1 se resume la evolución histórica en la definición de los síndromes mieloproliferativos.
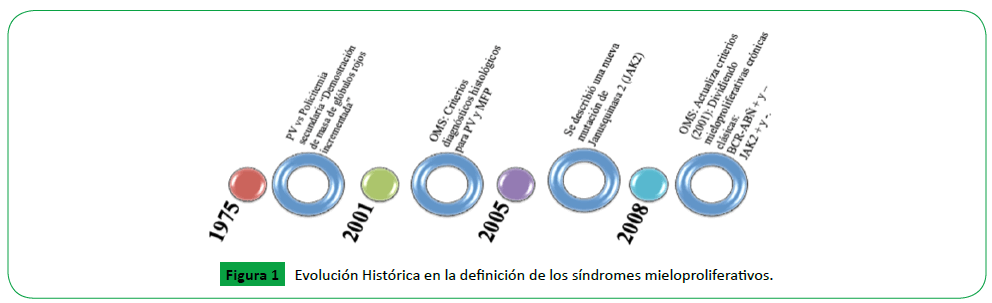
Figure 1: Evolución Histórica en la definición de los síndromes mieloproliferativos.
Las Neoplasias mieloproliferativas (NMP), son trastornos clónales que surgen de células madre hematopoyéticas afectadas por mutaciones somáticas que causan la producción anormal de células mieloides maduras [18]. Las NMP con cromosoma negativo de Philadelphia, incluyen policitemia vera (PV), trombocitemia esencial (TE) y mielofibrosis primaria, son las NMP más comunes; en cambio, la leucemia mieloide crónica (LMC) presenta por lo general cromosoma de Philadelphia positivo [9]. Estos trastornos se caracterizan por diversos grados de leucocitosis o trombocitosis; los pacientes con PV generalmente tienen un hematocrito marcadamente elevado [19]. Particularmente en PV y TE, la trombosis o la hemorragia representan un alto porcentaje de la morbilidad asociada de los trastornos, siendo la trombosis la más común [20]. Se estima que cada uno tiene una tasa de 0.5 a 2.5 por 100,000 personas por año. Así también como la mutación del JAK2 V617F ocurre en aproximadamente el 95% de los pacientes con PV, pero también en aproximadamente el 50% de aquellos con TE o PMF [21].
Policitemia vera
A Vaquez y Osler se les atribuye la descripción inicial de PV, como “proceso eritrocítico primario”, en 1892 y 1903, respectivamente. Las cifras de incidencia reportadas en PV varían de aproximadamente 0.5 a 2.6 por 100,000 habitantes [22]. Se ha sugerido una mayor incidencia de la enfermedad en personas de ascendencia judía. La edad media al momento del diagnóstico de PV es de aproximadamente 60 años con una ligera preponderancia masculina (1,2: 1) [23]. Aproximadamente el 7% de los pacientes son diagnosticados antes de los 40 años de edad.
PV es una enfermedad de células madre clonal con afectación mieloide trilinaje [24]. Además, algunos estudios sugieren heterogeneidad clonal, incluida la afectación clonal de linfocitos B. Además, las células progenitoras eritroides en PV muestran hipersensibilidad al factor de crecimiento de eritropoyetina (EPO), factor de crecimiento similar a la insulina (IGF) -1 y otras citoquinas [22].
La PV tiene una fase latente, proliferativa y agotada. El paciente generalmente acude a la consulta médica debido a los síntomas asociados con el aumento del volumen sanguíneo y la viscosidad o la función plaquetaria alterada: Deterioro de la circulación cerebral que produce dolor de cabeza, vértigo, visión borrosa, mareos, ataque isquémico transitorio, accidente cerebrovascular, fatiga, poca tolerancia al ejercicio, prurito, particularmente después del baño (causado por la sobreproducción de histamina), sangrado: epistaxis, hemorragia gastrointestinal superior (mayor incidencia de enfermedad de úlcera péptica), incomodidad abdominal por esplenomegalia y hepatomegalia, hiperuricemia puede causar nefrolitiasis y artritis gotosa [24-28]. Hasta el 40% de los pacientes experimentan trombosis arterial o venosa durante el curso de su enfermedad. Los eventos trombóticos cerebrales y esplácnicos ocurren también comúnmente [29].
Los principales criterios diagnósticos son: Hemoglobina mayor a 18.5 g/dL en hombres, o mayor a 16.5 g/dL en mujeres, u otra evidencia de aumento en el volumen de glóbulos rojos y presencia de mutación JAK2 V617F u otra mutación funcionalmente similar como JAK2 mutación del exón 12 [13-22]. Los criterios diagnósticos propuestos por la OMS para el diagnóstico de la policitemia vera se resumen en la Figura 2.

Figure 2: Criterios propuestos por la OMS para el diagnóstico de Policitemia Vera.
Más del 95% de los pacientes con PV portan la mutación JAK2 V617F; por lo que es más efectivo iniciar el estudio de un paciente con sospecha de PV con detección de mutación en sangre periférica para JAK2 V617F [11-12]. Por otra parte, para reducir al mínimo las consecuencias de falsos positivos o falsos negativos de los resultados de pruebas moleculares, así como la captura de los pocos casos de PV que son JAK2 V617F negativos, se debería usar la medición simultánea de nivel de eritropoyetina (EPO) en suero [7,8,26]. Si los resultados de ambas pruebas son sugestivos de PV (es decir, mutación positiva y baja EPO sérica), entonces el diagnóstico es probable y se recomienda el examen de médula ósea, pero no es esencial para realizar el diagnóstica [26]. Si el JAK2 V617F y los resultados de la prueba EPO sérica no son consistentes con el diagnóstico de PV (es decir, mutación negativa y EPO normal o aumentada), entonces no se recomienda una mayor investigación para PV a menos que el escenario clínico lo dicte de otra manera [11-12].
Esta mutación JAK2 V617F, es una mutación somática Guanina a Timina (G-a-T), en el nucleótido 1849, en el exón 14, que da como resultado la sustitución de valina por fenilalanina en el codón 617 [30], Y las mutaciones JAK2 del exón 12 (F537-K539delinsL, H538QK539L, K539L, N542-E543del) que incluyen tanto delecciones en marco, como mutaciones en el punto tándem [31] Inducen una proliferación independiente de citoquinas / hipersensible en líneas celulares que expresan el receptor EPO [11-12].
Se ha demostrado que el tratamiento específico de la PV influye positivamente en el riesgo de complicaciones macrovasculares y microvasculares, pero no en la evolución clonal a la mielofibrosis o la leucemia mieloide aguda (LMA post-PV) [32]. La flebotomía es la piedra angular de la terapia en PV y es la única modalidad de tratamiento que ha mejorado la supervivencia en pacientes afectados [33]; Se informa que la supervivencia media puede ser tan baja como 2 años en ausencia de dicho tratamiento [24,25,33]. Basado en estudios retrospectivos limitados en PV que mostraron un aumento progresivo en la incidencia de episodios oclusivos vasculares por encima de un nivel de hematocrito de 44% [33,34]. Así como otros estudios que mostraron un flujo sanguíneo cerebral subóptimo en rangos de valores de hematocrito entre 46% y 52%, el nivel de hematocrito diana terapéutico se ha establecido, durante mucho tiempo, en 45% o menos [35]. Además, debido a la diferencia fisiológica en los valores de hematocrito entre los dos géneros, así como entre diferentes razas, es razonable, aunque no basado en evidencia, apuntar a un nivel de hematocrito incluso más bajo (es decir, 42%) en mujeres y afroamericanos [33-35].
Trombocitemia esencial
La trombocitemia esencial (TE) fue descrita en 1934 por Emil Epstein y Alfred [36]. En 1981 Philip Fialkow usando el polimorfismo de la glucosa-6-fosfatodeshidrogenasa estableció la naturaleza clonal de los SMP y la confirmó [37]. Entre los síndromes mieloproliferativos, la TE es el más recientemente descrito [36]. Las cifras de incidencia informadas varían de 0.2 a 2.5 por 100,000 habitantes, con una mediana de edad al momento del diagnóstico de 60 años, aproximadamente el 20% de los pacientes con TE se diagnostica antes de los 40 años y en el grupo de pacientes de edad joven, la incidencia es más alta en las mujeres que en los hombres [37].
La base clonal de la TE se destacó por el análisis de la mutación JAK2 V617F que reveló la presencia de la mutación incluso en aquellos pacientes que presentan hematopoyesis "policlonal" mediante estudios de clonación ligados al cromosoma X [7,8]. Sin embargo, el evento clonogénico primario en TE permanece indefinido a pesar de las descripciones recientes de dos mutaciones de ganancia de función que ocurren en 50% (JAK2 V617F) y 1% (MPL W515L/K) de pacientes con TE, respectivamente [11-12].
En la actualidad, se cree que la diátesis hemorrágica en TE implica un síndrome de von Willebrand adquirido que se vuelve aparente en presencia de trombocitosis extrema [38].
El 50% de los pacientes con TE son asintomáticos al diagnóstico y la trombocitosis aparece como un hallazgo en un hemograma de rutina [19,39]. El resto se presenta con síntomas vasomotores por obstrucción de la microcirculación o trombosis y/o hemorragia de magnitud variable [39]. La obstrucción de la microcirculación es frecuente: fenómeno de Raynaud, isquemia acrocianótica que puede evolucionar a una gangrena periférica, disturbios visuales y auditivos, cefaleas, mareos y otros síntomas neurológicos isquémicos transitorios, como escotomas centellantes, disartria, inestabilidad motora, ceguera monocular y paresias o hemiparesias transitorias [11,12].
Aunque la trombocitosis (Recuento plaquetario > 450 x109 y sostenido) es el sello distintivo de la TE, más del 85% de los casos con trombocitosis observados en la práctica clínica habitual son reactivos (trombocitosis secundaria) y se asocian con otras enfermedades comórbidas [40]. El grado de trombocitosis es un discriminador pobre para diferenciar TE de la trombocitosis secundaria. Sin embargo, el escenario clínico a menudo es útil para distinguirlas [36]. En la práctica clínica habitual, es importante excluir la contribución de la anemia por deficiencia de hierro o un estado hipoesplénico como posibles causas de una trombocitosis [40]. La posibilidad de una trombocitosis secundaria asociada a un proceso inflamatorio o maligno oculto se aborda mediante la medición de la proteína C reactiva (PCR) u otros reactivos de fase aguda. Como se ha demostrado en los estudios diagnósticos, una TE sin complicaciones debe ir acompañada de ferritina sérica normal, frotis periférico casi sin importancia y una PCR en suero normal [40,41].
A diferencia del caso de PV, la utilidad de la detección de mutación para JAK2 V617F para el diagnóstico de TE está limitada por un valor predictivo negativo subóptimo y la falta de especificidad diagnóstica en el contexto de neoplasias mieloides [7,8,15]; Cabe señalar que solo la mitad de los pacientes con TE portan JAK2 V617F y que la presencia de la mutación no puede diferenciar TE de otra NMP. Por lo tanto, aunque la detección de sangre periférica para JAK2 V617F ayuda a agilizar más investigaciones, a menudo se requiere una biopsia de médula ósea para ayudar con el diagnóstico diferencial de la trombocitosis primaria [11]. En general, la presencia de JAK2 V617F argumenta contra la posibilidad de un proceso no clonal, como infección o inflamación, o una malignidad no mieloide como el linfoma o el cáncer metastásico [11,12,37]. Los criterios propuestos por la OMS para el diagnóstico de la Trombocitemia esencial se resumen en la Figura 3.

Figure 3: Los criterios propuestos por la OMS para el diagnóstico de la Trombocitemia esencial.
La TE se trata eficazmente con dosis bajas de aspirina (81 a 325 mg/día) [41,42]. Sin embargo, es importante excluir la posibilidad de una enfermedad de von Willebrand adquirida clínicamente significativa (actividad de cofactor de ristocetina de <30%) antes de usar aspirina en los pacientes con recuentos de plaquetas superiores a 1 millón de células/μL [42,43].
Mielofibrosis primaria
La MFP fue descrita por primera vez en 1879 [36]. El curso clínico de la PMF se caracteriza por anemia progresiva, hepatoesplenomegalia marcada, caquexia, desarrollo de hematopoyesis extramedular no hepatoesplénica y evolución a LMA [44,45].
Similar a la situación en PV y TE, la lesión genética que inicia la enfermedad en MFP no se ha identificado, y el diagnóstico actual se basa en características clínicas y de laboratorio características, pero no específicas, que incluyen fibrosis de médula ósea [46]. Además de la mieloproliferación clonal, la médula ósea en MFP podría mostrar un exceso de fibrosis de colágeno, osteosclerosis y angiogénesis [47]. Estos cambios se han asociado con alteraciones en los niveles celulares y extracelulares de diversas citoquinas fibrogénicas y angiogénicas, incluido el factor de crecimiento transformante β (TGF-β), factor de crecimiento de fibroblastos básico y factor de crecimiento derivado de plaquetas [46-48]; sin embargo, no se han identificado las mutaciones clonogénicas primarias, aunque se ha prestado mucha atención a las mutaciones de ganancia de función descritas que implican la tirosincinasa JAK2 (JAK2 V617F) y el receptor de trombopoyetina (TPO) (MPL W515L / K), pero la mutación del MPL W515L/K parece ser específico para MFP o TE, aunque la frecuencia mutacional es sustancialmente menor (<5) [11,12].
Para el diagnostico, en la Medula ósea (MO) la presencia de proliferación de megacariocitos y atipia, generalmente acompañada de reticulina y/o fibrosis de colágeno, o, en ausencia de fibrosis reticulínica significativa, los cambios en los megacariocitos deben ir acompañados de una mayor celularidad de la MO caracterizada por proliferación granulocítica y, a menudo, disminución de la eritropoyesis [48]. Los criterios propuestos por la OMS para el diagnóstico de la MFP se resumen en la Figura 4.

Figure 4: Los criterios propuestos por la OMS para el diagnóstico de la MFP.
No debe cumplir con los criterios de la OMS para PV, LMC, síndrome mielodisplásico u otra neoplasia mieloide [47] Se debe tener especial precaución para no diagnosticar erróneamente leucemia mieloide crónica (LMC) o leucemia de células pilosas como una MFP. Se abordan estos problemas y se incorpora el cribado de la mutación JAK2 V617F en los criterios diagnósticos de la OMS [44]. Los datos pronósticos más recientes sugieren una supervivencia inferior en MFP asociada para el haplotipo JAK2 46/1, carga baja del alelo JAK2 V617F, o presencia de mutación del gen de Isocitrato deshidrogenasa (IDH) [11,12,47,49,50].
Conclusion
Los síndromes mieloproliferativos crónicos (SMC) JAK2+, que comprenden PV, TE y MFP, son cánceres hematopoyéticos clonales que tienen un curso indolente. Las manifestaciones clínicas de estas entidades se superponen, al igual que sus impulsores genéticos. Además del mimetismo fenotípico, cada tipo de neoplasia mieloproliferativa es capaz de evolucionar hacia otro tipo, lo que dificulta el diagnóstico, la evaluación de riesgos y las opciones terapéuticas.
Es importante evaluar la presencia de la mutación JAK2 en pacientes con SMC, y en aquellos pacientes que no han respondido de manera óptima a la terapia primaria dirigida a la enfermedad.
La secuenciación completa de genes de pacientes con cánceres de sangre es cada vez más accesible y rutinaria. El desafío actual es la integración de datos clínicos con el perfil del genoma de diagnóstico puede proporcionar predicciones de pronóstico que se adaptan a los pacientes de manera individual y dicha información empoderará al médico y respaldará decisiones complejas sobre la elección y la intensidad de la terapia, el reclutamiento para los ensayos clínicos y la perspectiva clínica a largo plazo.
33214
References
- Ferré OJ, Sánchez-Guijo F (2016) Síndromes mieloproliferativos. Medicine - Programa de Formación Médica Continuada Acreditado. 12: 1213-1223.
- Kutti J, Ridell B (2001) Epidemiology of the myeloproliferative disorders: Essential thrombocythaemia, polycythaemia vera and idiopathic myelofibrosis. Pathologie Biologie 49: 164-166.
- Mengual FB, Pérez JT, Sesé GM, Pérez MF, Cardona RB (2013) Paciente terminal. Guía de actuación clínica en ap España.
- Pascual López L, Pastor Domenech V, Gutiérrez Valverde J, Nieto Giménez, F (2000) La atención al paciente con cáncer en fase terminal en sus últimos días de vida (I). Revista Valenciana Medicina de Familia 8: 17-24.
- Pardo C, de Vries E, Buitrago L, Gamboa O (2017) Atlas de mortalidad por cáncer en Colombia. Revista Instituto Nacional de Cancerología Cuarta edición. Bogotá DC 1: 124.
- Swerdlow SH, Campo E, Lee Harris N, Jaffe E, Pileri S, et al. (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. IARC (4th ed.) Lyon, Francia.
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, et al. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. The Lancet 365: 1054-1061.
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, et al. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7: 387-397.
- Pérez MJ, Acevedo TP (2013) Epigenética: una nueva herramienta para el estudio de la leucemia mieloide crónica. Medicina & Laboratorio. 19: 243-255.
- Vardiman JW, Harris NL, Brunning RD (2002) The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100: 2292-2302.
- Gorbenko AS, Stolyar MA, Olkhovskiy IA, Vasiliev EV, Mikhalev MA (2019) Parallel algorithm for myeloproliferative neoplasms testing: the frequency of double mutations is found in the JAK2/MPL genes more often than the JAK2/CALR genes, but is there a clinical benefit? Clinical Chemistry and Laboratory Medicine 57: e60-e62.
- Li ZL, Gao L, Zhang H, Zhang CX, Chen YR, et al. (2018) Detection and Diagnostic Values of JAK2, CALR, MPL Gene Mutations in 208 Cases of BCR/ABL1 Negative Chronic Myeloproliferative Diseases. Zhongguo Shi Yan Xue Ye Xue Za Zhi 26: 1122-1128.
- Turkington RC, Arnold EC, Percy MJ, Ranaghan LA, Cuthbert RJ, et al. (2007) Comparison of diagnostic criteria for polycythaemia vera. Hematology 12: 123-130.
- Ancochea Serra A (2018) Reproducibilidad de los criterios de la OMS en el diagnóstico de la policitemia vera. Tesis doctoral. Universitat autónoma de Barcelona.
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain of function mutation in JAK2 is frequently found in patients with myeloproliferative disorders. N Engl J Med 2005; 352: 1779-90.
- Murphy S, Iland H, Rosenthal D, Laszlo J (1986) Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol 23: 177–182.
- Tefferi A, Thiele J, Vardiman JW (2009) The 2008 World Health Organization classification system for myeloproliferative neoplasms: Order out of chaos. Cancer: Interdisciplinary International Journal of the American Cancer Society 115: 3842-3847.
- Vannucchi AM, Guglielmelli P, Tefferi A (2009) Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin 59: 171-191.
- Tefferi A, Barbui T (2015) Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk‐stratification and management. American Journal of Hematology 90: 162-173.
- Passamonti F, Rumi E, Pungolino E, Malabarba L, Bertazzoni P, et al. (2004) Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. The American Journal of Medicine 117: 755-761.
- Daniela L, Pablo M, Andreína B, Natalia T, Silvia P (2007) Determinación de la mutación V617F del gen JAK2 en los síndromes mieloproliferativos crónicos en nuestro país: A propósito de un caso. Rev Méd Urug 23: 122-125.
- Dawson MA, Huntly BJ (2013) The pathogenesis, diagnosis, and treatment of polycythaemia vera. Neoplastic Diseases of the Blood, Springer, New York, pp: 135-153.
- Johansson P (2006) Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. In Seminars in Thrombosis and Hemostasis 32: 171-173.
- Spivak JL (2002) Polycythemia vera: myths, mechanisms, and management. Blood, The Journal of the American Society of Hematology 100: 4272-4290.
- Díaz Sánchez OR (2019) Importancia del diagnóstico precoz de la policitemia vera: Experiencia en un centro.
- López AA, Placeres LL, Méndez IMS, Santos YB (2020) Caracterización clínico-laboratorial de las neoplasias mieloproliferativas crónicas durante diez años en San Cristóbal. 16 de Abril 59: 888.
- de la Torre RG, Morán MT, Caro JM, Simón JF, de la Campa JA (2009) Fiebre y dolor abdominal en la policitemia vera. Revista Clinica Espanola 209: 354-356.
- Martínez-Flores JL, Ramos-Peñafiel CO, Santoyo-Sánchez A, Kassack-Ipiña JJ, Gallardo-Trillanes E, Castellanos-Sinco H, et al. (2016) Implicaciones clínicas y de pronóstico de la mutación JAK2 V617F en pacientes con neoplasias mieloproliferativas crónicas. Revista de Hematología 17: 161-168.
- De Stefano V, Za T, Rossi E, Vannucchi AM, Ruggeri M, et al. (2008) Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 93: 372-380.
- Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, et al. (2005) Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 106: 2162-2168.
- Sieza Y, Di Camilo I, Mazziott L, Archuby ML, Riva ME, et al. (2018) Distribución de mutaciones en JAK2, MPL y CALR en pacientes con sospecha de neoplasias mieloproliferativas crónicas Phi negativas provenientes de hospitales públicos de la provincia de Buenos Aires. Hematología 22: 151-156.
- Martínez Avilés L (2015) Clonalidad hematopoyética en la policitemia vera y la trombocitemia esencial y su papel en la transformación mielofibrótica. Tesis doctoral. Universitat Autònoma de Barcelona.
- Barbui T, Passamonti F, Accorsi P, Pane F, Vannucchi AM, et al. (2018) Evidence-and consensus-based recommendations for phlebotomy in polycythemia vera. Leukemia 32: 2077-2081.
- Abello V, Quintero G, Espinosa D, Solano MH, Casas CP, et al. (2017) Descripción de las caracteristicas clínicas de las neoplasias mieloproliferativas crónicas (NMPC). Primer informe del registro colombiano de NMPC. Acta Medica Colombiana 42: 35-41.
- Lozano JC, Casas CP, Abello V, Solano MH (2012) Características clínicas y paraclínicas de las neoplasias mieloproliferativas crónicas cromosoma Filadelfia negativas. Acta Médica Colombiana 37: 66-73.
- Jiménez SI (2017) Neoplasias mieloproliferativas. De la clínica a la biología molecular. Acta Médica Colombiana 42: 15-17.
- Sociedad Argentina de Hematología (2017) Guías de Diagnóstico y Tratamiento: Año 2017.
- Parnes A, Ravi A (2016) Polycythemia and thrombocytosis. Primary Care: Clinics in Office Practice 43: 589-605.
- Gisslinger H (2006) Update on diagnosis and management of TE. Semin Thromb Hemost 32: 430-436.
- Álvarez JF, Bedoya-Trujillo N, Saldaña J (2018) Enfoque clínico de la trombocitosis, una revisión de la literatura. Salutem Scientia Spiritus 4: 41-48.
- Sulai NH, Tefferi A (2012) Why Does My Patient Have Thrombocytosis? Hematol Oncol Clin North Am 26: 285-301.
- Rizzo J (2018) Evaluation of aspirin responsiveness in essential thrombocythemia patients.
- Kornblihtt LI, Vassallu PS, Heller P, Molinas FC (2002) Diez años de experiencia con anagrelide en el tratamiento de la trombocitemia esencial. Medicina 62: 231-236.
- Lorena MO, Alejandra AS, Juan DGE, Federico RWL (2017) Mielofibrosis. Acta méd. Grupo Ángeles 15: 225-229.
- Tefferi A, Lasho TL, Jimma T, Finke CM, Gangat N, et al. (2012) One thousand patients with primary myelofibrosis: The mayo clinic experience. Mayo Clinic Proceedings. 87: 25-33).
- Tefferi A (2000) Myelofibrosis with myeloid metaplasia. New England Journal of Medicine 342: 1255-1265.
- Reilly JT, McMullin MF, Beer PA, Butt N, Conneally E, Duncombe A, Et al. (2012) Guideline for the diagnosis and management of myelofibrosis. Br J Haematol 158: 453-471.
- Arora B, Ho CL, Hoyer JD, Mesa RA, Tefferi A (2004) Bone marrow angiogenesis and its clinical correlates in myelofibrosis with myeloid metaplasia. Haematologica 89: 1454-1458.
- Tefferi A (2010) Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 24: 1128–1138.
- Castillo I, Ojea MA, Boqué C, Asensio A, Hermosilla MM, et al. (2013) Efectividad y seguridad de lenalidomida en pacientes con mielofibrosis: serie de casos del programa de uso compasivo español. Farm Hosp 37: 135-142.

