Keywords
Sickle cell anaemia; Haemoglobin S; Hydroxyurea; Gene therapy; Genetics
Introduction
Sickle Cell Anaemia (SCA), the most common type of Sickle Cell Disease (SCD) is believed to be a multisystem, monogenic heritable blood disorder, often associated with occurrences of acute illness and progressive organ damage [1,2]. The disease is characterised by abnormally shaped Red Blood Cells (RBCs), which are removed from circulation and destroyed at increased rates, leading to anaemia. In addition to the aforementioned clinical features, the distorted RBCs also cause vascular occlusion, leading to haemolysis and tissue infarction. The first published account of SCD was described in a dental student in 1910 who was presented with pulmonary symptoms [3]. Henrick was the first to coin the term “sickle-shaped” to describe thecharacteristic appearance of the erythrocytes of the patient
(Figure 1) [3].
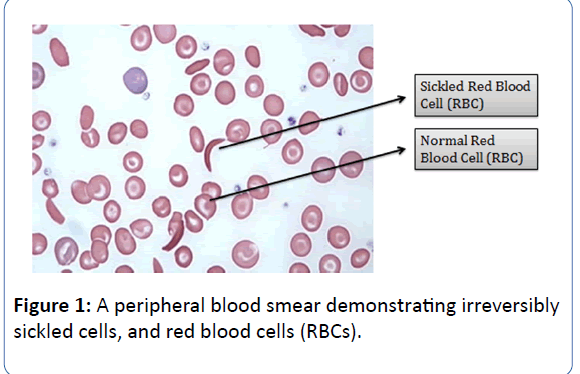
Figure 1: A peripheral blood smear demonstrating irreversibly sickled cells, and red blood cells (RBCs).
Studies by Pauling and colleagues [4] later determined that a point mutation in the gene coding the β-chain of the haemoglobin molecule resulted in a single amino acid substitution, thereby resulting in sickle cell haemoglobin or HbS. The structure of haemoglobin contains four protein subunits, of which two are β-globin and the remaining two are α-globin subunits. Specifically, β-globin is a larger protein subunit of haemoglobin compared to α-globin. Haemoglobin S (HbS) replaces both the β-globin subunits, and sickle cell anaemia occurs when the patient possesses two copies of the sickle cell genes (HbSS). Later, in 1956 Vernon Ingram and colleagues identified the anomaly in the amino acid sequence and described the replacement of amino acid-glutamic acid by valine at 6th position in β-globin protein subunit [5,6]. The sustitution causes the abnormal haemoglobin S subunits to form aggregates with other haemoglobin molecules within the RBC. These molecules then adhere together and develop into long and rigid chains which distort the RBC into a characteristic sickle shape, thereby leading to blockage of blood vessels and finally impair blood flow through vessels [7]. This paves the way for a cascade of complications like anaemia, hypoxia, fatigue, delayed growth etc. Additionally, the increased breakdown of RBC can cause yellowing of eyes and skin, showing signs of jaundice and the lack of oxygen rich blood results in organ damage of the spleen, liver, lungs, kidney, especially the brain which can also result in stroke [8].
SCA is a prevalent single gene disorder among black Americans affecting nearly 0.003% black Americans in the United States. Additionally, an approximate of 15% of the black population in the United States is afflicted with SCA. The disease is prevalent among the Senegal, Benin, Bantu and Cameroon tribes of Arica and Arabian-Indian tribes of the Asian haplotype [9]. More than 305,000 infants are conceived every year with SCD for the most part in low and middle income nations, and around 75% of these births occur in sub-Saharan Africa, representing an expanding wellbeing burden and adding to early adolescence mortality [10]. Every year, 275,000 youngsters with sickle cell weakness are conceived on the planet, primarily in Africa, where around 1.5% of babies have the sickness. In corroboration of this, Nigeria has the largest population of people with SCD in the world with over 150,000 babies born with this serious condition every year [11].
This review discusses the pathophysiology and genetics of the disease in addition to illustrating the various manifestations. Furthermore, a widespread account of the advancements in therapy and future prospective of treatment has been discussed.
Genetics of SCA
Pioneering studies leading to the discovery of Haemoglobin S (HbS) by Linus Pauling and colleagues in 1949 demonstrated for the first time that the production of an abnormal protein could be the cause of a genetic disorder [4]. It was during the same year, that studies revealed an autosomal recessive model of inheritance of the disease [12]. The iron-containing metalloprotein, Haemoglobin (Hb), encoded by the HBB gene, in the mammalian red blood cells (RBCs), responsible for transport of oxygen to the tissues is known to change structure during human development [13]. Broadly classifying, two components exist in adults, namely: major haemoglobin (HbA), and minor haemoglobin A2 (HbA2). The bulk of the haemoglobin during advanced foetal life is foetal haemoglobin (HbF). Each of these haemoglobins is composed of two pairs of dissimilar globin chains. The adult haemoglobins and foetal haemoglobin have α chains combined with β (HbA, α2β2), δ (HbA2, α2δ2), or γ chains (HbF F, α 2γ2) [13]. Studies have revealed that genetic changes encircling mutations in the haemoglobin gene (HBB) which codes for the protein haemoglobin appears to be the causative rationale for the widespread occurrence of SCA [14]. Additionally, the HBB gene is known to provide instructions for creating the beta-globin (β-globin), the various versions of which are a result of diverse mutations in the HBB gene [14].
A normal molecule of haemoglobin A (HbA) predominates as the adult human haemoglobin tetramer consisting of four globin subunits, typically two α chains and two β chains [15]. Two normal globin chains of α and β constitute the normal structural composition of HbA, while anomalies in the structural components of the β-globin chain often leads to structural malformations thereby inducing the production of defective haemoglobin [15,16]. Briefly, certain mutations in the HBB gene produce an abnormal version of β-globin known as Haemoglobin S (HbS), while other mutations result in additional anomalous forms, namely Haemoglobin C (HbC) and Haemoglobin E (HbE) [14]. In individuals’ diagnosed with SCD, at least one of the β- globin subunits is replaced with HbS. However, in SCA, known to be the most common form of SCD, both the β-globin subunits of haemoglobin are replaced by two copies of the variant haemoglobin S (HbSS) resulting in the production of sickled haemoglobin in the red blood cells (RBCs) [6,14]. In other variants of SCD, individuals are often heterozygotes wherein, they possess a copy of the HbS variant and another β- globin namely, HbC or haemoglobin β-thalassemia (H β-thal) [6].
Pattern of inheritance of SCA
SCA is an autosomal recessive gene disorder [17]. The parents of an individual with an autosomal recessive condition carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. The health of the parent is rarely affected, but they have one mutated gene (recessive gene) and one normal gene (dominant gene) which leads to the defective condition in the offspring. If the recessive genes from both the parents fuse together, the symptoms of SCD are observed in the offspring (Figure 2) [17]. It follows an autosomal recessive pattern of inheritance, generating a phenotypic ratio of 1:2:1 (Unaffected child: Unaffected carrier child: Affected child). The heterozygous mutated gene carrying individual is the carrier for SCA.
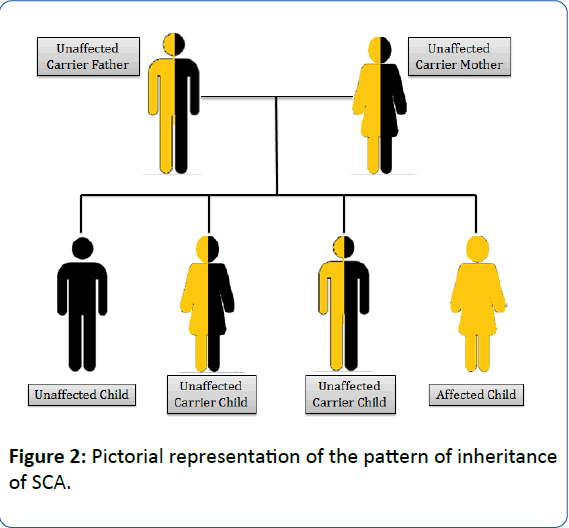
Figure 2: Pictorial representation of the pattern of inheritance of SCA.
Molecular Pathophysiology
In the course of the process of deoxygenation which follows the passage of RBCs in the microcirculation, the Hb molecule undergoes a conformational change which requires the packaging of high concentration of Hb molecules in RBCs to be extraordinarily soluble [18,19]. SCA occurs due to a single nucleotide polymorphism (SNP) in the short arm of chromosome 11 [20]. In SCA, the presence of HbS results in replacement of the hydrophilic glutamic acid with the hydrophobic amino acid valine at position 6 of the short arm of chromosome 11 of the β- globin chain. This results in a hydrophobic interaction between the haemoglobin molecules, thereby triggering the formation of a large polymer [15,18,19]. The process of polymerization of deoxygenated HbS forms the essential and primary event in the molecular pathogenesis of the disease. This further induces the formation of helical, twisted, rope-like fibres which when grouped together stiffen and distort the shape of RBC into a variety of elongated shapes, decreasing the deformability and conferring it a characteristic “sickle shape” [15,18,21]. The process of polymerization triggers a cascade of several cellular and molecular events, ultimately leading to vascular obstruction and haemolysis which participate in the overall molecular pathophysiological mechanism of the disease (Figure 3) [19]. The series of events following polymerization results in a nonselective increase in membrane cation permeability to sodium, potassium, calcium and magnesium, which upon accumulating in the RBCs, activate several cell membrane transport systems resulting in a cumulative discharge of water causing the RBCs to dehydrate and become dense. Consequently, the concentration of haemoglobin in the cell increases, in turn accelerating the process of polymerization which leads to added dehydration. Such alternate episode of reversible polymerization and irreversible dehydration forms a dense, irreversible sickled RBC which appears distorted in shape and contributes to vasoocclusion and haemolysis [21-23].
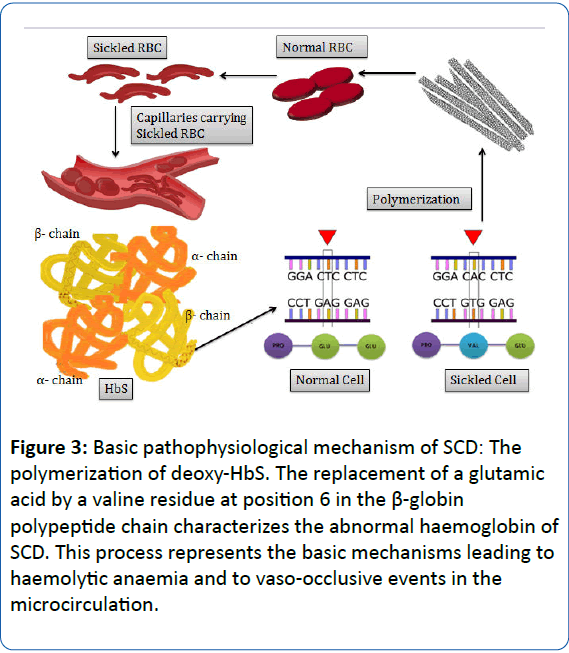
Figure 3: Basic pathophysiological mechanism of SCD: The polymerization of deoxy-HbS. The replacement of a glutamic acid by a valine residue at position 6 in the β-globin polypeptide chain characterizes the abnormal haemoglobin of SCD. This process represents the basic mechanisms leading to haemolytic anaemia and to vaso-occlusive events in the microcirculation.
The elucidation of the molecular mechanisms of SCD remains an unparalleled model, and marks a remarkable achievement in itself. However, factors and processes such as vaso-occlusion and haemolysis require further illustration.
Diseases Associated with SCA
Paleness is a typical condition in principally sick patients and results in a high prerequisite for blood transfusions related with poor results [24]. Anaemia is the primary symptom which is associated with SCA. The adaptability of the erythrocytes is crumbled, its life expectancy is abbreviated, and the capillaries in the body are most likely going to impede. This implies that there are insufficient solid red platelets to transport oxygen all through the body. When this happens, a person might present with tiredness, pale skin colour, and delayed puberty. Pain is the most widely recognized entanglement of SCD, and typically most common reason that the patients with SCD go through the crisis. In the sequential effect of SCA, RBCs of most of the patient loose affinity for oxygen and the patient suffers with hypoxia. The patient becomes fatigued easily and experiences shortness of breath.
Acute chest syndrome (ACS), described by fever and new opacification of the lungs on chest radiograph, is the second most common reason for hospitalization in patients with sickle cell ailment and causes 25% of deaths. Rehashed scenes of ACS are related with advancement of incessant lung sickness, with dynamic hypoxemia and diminished survival [25]. About 30% of adult patients with SCA suffer from pulmonary hypertension, and the complication is a strong negative prognostic factor. Recent examinations have uncovered that adenosine concentrations are fundamentally lifted in SCD and add to infection pathology by initiating adenosine receptors on red platelets. Apart from adenosine, hypoxia additionally causes haemoglobin discharge through haemolysis [26].
SCA influences vast majority of tissues in the body. SCA brings about both anatomical adjustments and aggravations in renal capacity. Majority of the sickle cell iron deficient patients have enormous kidneys, which may be attributed to expanded renal blood volume [27]. The imperfection in concentrating urine capacity in sickle cell trait is accepted to be the result of intracellular polymerization of HbS in erythrocytes that causes impediment of tiny vessels in the vasa recta of the renal medulla. The seriousness of the deformity in concentration of urine may be connected to the level of sickling process of erythrocytes [28]. During later stages of this disorder, renal dysfunction results in sickle nephropathy.
In a severe situation, large number of sickle cells gets trapped in the spleen and makes it enlarged. Symptoms shown by the individual are extreme thirst, left abdominal pain, weakness, pale lips, fast heartbeat and breathing. This event can be life threatening, in most cases. Medically, this discomfort is known as splenic Sequestration [29].
In the worst case scenario, major organ failure is the critical situation which arises. Initially, clinically apparent as autosplenectomy, followed by cerebral infarction during childhood and finally in young adulthood as end stage renal failure (glomerulosclerosis), disabling leg ulcers, generalized osteonecrosis, intracranial haemorrhage, retinopathy and sickle chronic lung disease [30].
Diagnosis of SCA
Prenatal screening
Currently it is feasible for specialists to analyse sickle cell ailment before birth. This can be accomplished by utilizing a specimen of amniotic fluid (liquid in the sac encompassing a developing foetus) or tissue taken from the placenta. This test should be possible in the initial couple of long stretches of pregnancy. Pre-birth conclusion of sickle cell iron deficiency is conceivable utilizing amniocentesis or chorionic villus sampling. The specimen received is then tried for DNA investigation of the foetal cells. Testing before birth should be possible as early as first trimester of pregnancy. This testing examines for the sickle haemoglobin gene instead of the abnormal haemoglobin [31].
New-born screening
In the United States, the District of Columbia, and the U.S. domains require that each child is tried for SCD as a component of an infant screening program. It is essential to analyse SCD right on time to avoid entanglements. In infant screening programs, blood from a foot rear area prick is gathered in "spots" on a unique paper. The haemoglobin from this blood is then dissected in exceptional labs. Results will then be discussed with the parents. If the test shows sickle haemoglobin, a second blood test is done to confirm the diagnosis [32].
It is noteworthy that, when a child has sickle cell trait or SCD, a future sibling, or the child’s own future may be at risk. These possibilities should be discussed with the primary care doctor, haematologist, and/or genetic counsellor [33].
For young individuals displaying symptoms of SCA, blood smear is observed under microscope to check the abnormal sickle-shaped cells. Smearing of blood using a special lowoxygen preparation is done for testing and this is referred to as sickle prep. Other prep tests can likewise be utilized to distinguish the strange HbS. Preparatory tests also include dissolvability tests performed on the blood samples. The infection can be affirmed by particularly evaluating the kinds of haemoglobin, utilizing a haemoglobin electrophoresis test [34]. This electrophoresis test recognizes the haemoglobin in the blood by isolating them. The partition of the diverse haemoglobins is conceivable due to the unique electrical charges they have on their protein surfaces, causing them each to move distinctively in an electric field [35].
Treatment Procedures
Blood transfusions are utilized to treat extreme anaemia. A sudden exacerbation of weakness caused due to disease or augmentation of the spleen is a typical purpose behind a transfusion. Nonetheless, excess blood transfusions might cause medical issues as a result of the excess iron pumping in the blood. This over-burden, called haemosiderosis, can harm liver, heart, pancreas and different organs, prompting maladies, for example, diabetes mellitus. Press chelation treatment ought to be begun in patients with SCD accepting general blood transfusions to lessen abundant press levels.
SCD is a genetic disorder endemic in malaria prone areas. Therefore, the need for prompt initiation of efficacious antimalarial therapy in patients with SCD is of utmost importance. In a randomized controlled trial, with 60 children with SCD and acute uncomplicated malaria were treated with artesunate-amodiaquine (AA) also known as artemetherlumefantrine (AL). It was observed that AA and AL showed similar clinical and parasitological effects in the SCD and non- SCD groups. This could be attributed to alterations in WBC and platelet counts [36].
The goals of treating SCD are to prevent infections, relieve pain, prevent organ damage, strokes, treat anaemia and control complications [23].
Treating pain
The most difficult sign of SCA appears to be the pain. Mild pain is often treated with heating pads and counter medicines. In order to suppress the pain, fluids and pain-controlling medicines are administered. The body tends to become dehydrated due to pain, thus fluids are conferred either orally or intravenously. Severe pain may need to be treated in a hospital [37].
Common medicines used to treat pain crises include acetaminophen, narcotics and non-steroidal anti-inflammatory drugs (NSAIDs). Mild to moderate pain are usually treated with NSAIDs or acetaminophen. Moderate to severe pain is often treated with narcotics. The narcotic may be used alone or in combination with NSAIDs or acetaminophen. Certain counter pain medications available are aspirin and ibuprofen. In case of severe pain opioid (i.e. morphine) medications is given on a daily basis, along with additional pain medication [31].
Several parallel treatments have been used as combination or primary therapy for acute episodes of sickle cell pain. Nonsteroidal antiinflammatory agents such as ketorolac, piroxicam, and ibuprofen have been assessed for monotherapy and in combination with opiates for vasoocclusive crises associated with SCA [38]. A Nigerian trial found that piroxicam was as effective as aspirin for the treatment of vasoocclusive
crisis and had fewer side effects [39]. Furthermore, in a blinded, crossover trial, ketorolac provided superior pain control with fewer side effects than meperidine [40-42].
Prevention
HbF is a variant of Haemoglobin, which is found to impede the advance of HbS quality in an individual. In an examination, it was demonstrated that large amounts of HbF in children may diminish the seriousness of the sickness. This could be possible because of the hindrance of polymerization of HbS by HbF [43].
Hydroxyurea treatment might be useful in decreasing emergencies and the requirement for transfusions. Hydroxyurea does not cure sickle cell infection. It can incredibly lessen a portion of the complexities of the illness. It doesn't work effectively, if not taken as recommended. For the major part, it takes a while before any positive outcomes are observable. It is vital to recollect that hydroxyurea must be taken each day for it to function effectively. Those with extreme sickle cell iron deficiency may profit by daily consumption of hydroxyurea. Hydroxyurea is utilized to anticipate difficult pain but not to treat them [44].
Hydroxyurea treatment enables the red platelets to remain round and adaptable. This gives them a chance to move effortlessly through venules. To some extent, this happens because hydroxyurea expands the measure of foetal haemoglobin (HbF) in red platelets. As mentioned above, infants have HbF when they are born. HbF shields them from sickle cell complexities (medical issues) amid the first couple of months of life. With an expanded measure of HbF, red platelets are less inclined to change into sickle or banana shape. In most of us, the measure of HbF diminishes after the initial couple of long stretches of life. A few people have more HbF than others. Individuals with sickle cell disease who have more elevated amounts of HbF for the most part have less difficulties of the ailment [45]. The exact mechanism of activity of hydroxyurea isn't known in spite of the fact that the enlistment of HbF amalgamation and macrocytosis appear to be the possible mechanism [46]. Hydroxyurea was endorsed by the Food and Drug Administration (FDA) for the treatment of grown-ups with direct to extreme SCA and sickle-β-thalassemia [47].
The side effect of hydroxyurea includes myelotoxicity, which is reversible upon discontinuation of the medication. Opposite reactions that have been portrayed in animal studies considers its teratogenic and cancer-causing potential. Also, hydroxyurea has idiosyncratic reactions that happen in a few people and this might be a hereditary or epigenetic phenomenon. Studies report three idiosyncratic side effects of hydroxyurea in four patients with sickle cell iron deficiency (SS). Desquamating rash, diminished charisma and incomplete complex seizures have been already portrayed in patients who are hydroxyurea responders. In spite of the fact that these responses are uncommon, they can be not serious in few patients [48].
Preventing infections associated with SCA
Bacterial contaminations can be a noteworthy complexity of SCA, yet regularly they can be forestalled or treated. Children who have sickle cell illness hints at early onset of infections and diseases, for example, fever, trouble breathing, or restricted bone agony,
To avert diseases in infants and youthful kids, medications include:
• Daily dosages of penicillin: Treatment may start ahead of schedule at 2 months of age and proceed until the point when the kid is no less than 5 years of age.
• All routine immunizations (counting a yearly influenza shot), in addition to vaccination(s) against Streptococcus pneumonia.
Adults who have sickle cell ailment ought to likewise get influenza shots consistently and get inoculated against pneumococcal diseases. The two grown-ups and youngsters are in danger for an assortment of diseases, for example, pneumonia and bone contaminations. They ought to be inspected at whatever point they encounter fever since early finding and treatment result in better results [49].
Approaches and advancements in the therapy of SCA
The current understanding of the molecular pathophysiology of SCA has led to the development of several independent approaches and advancements in the therapy. A notable few of these theories, namely, pharmacologic induction of HbF, chemical inhibition of Hb polymerization and reduction of the intracellular haemoglobin concentration have been subjected to thorough laboratory and clinical investigations.
Pharmacological induction of HbF: Several biochemical, clinical, epidemiological and laboratory investigations have converged to support the notion that HbF is a very potent inhibitor of the deoxygenation of HbS in vitro [50]. Patients homozygous for SCA and belonging to the Bedouin area of Saudi Arabian Peninsula and central provinces of India, typically have relatively high amounts of HbF associated with relatively mild clinical manifestations of the disease [43,51-53,]. Based on these compelling biochemical and clinical evidence, it was proposed that the pharmacological inhibition of HbF production may act as an effective therapeutic strategy to benefit patients with SCA. Additionally, with studies elucidating the cloning of different globin genes and their mechanisms responsible for regulation gained importance in the late 1970s and 1980s [54-56]. It became very evident that epigenetic factors such as DNA methylation and histone acetylation were influential towards developmental regulation and the expression of globin genes [57-59]. This led to the theory that drugs which alter the epigenetic configuration of the γ-globin genes may furnish a viable therapeutic approach to the pharmacological induction of HbF, thereby influencing the inception of the first pharmaceutical agent, namely 5-azacytidine [53].
5-Azacytidine, an antineoplastic drug was the first agent used to induce HbF expression via epigenetic silencing of the γ-globin genes in adult life, the rationale for which was based on the discovery that the actively transcribed adult β-globin genes are hypomethylated and the non-transcribed foetal γ-globin genes are hypermethylated. Contrastingly, in foetal life, the adult β- globin genes are hypermethylated and the nontranscribed foetal γ-globin genes are hypomethylated [53,58,59]. Furthermore, in addition to inhibiting the methylation of DNA, it was observed to induce a marked increase in the levels of HbF in anaemic baboons [60]. Inspite of these promising results, limited clinical trials testing demonstrated the concerns regarding potential carcinogenicity of this drug in patients with SCA. Thus, focus was shifted towards other anti-tumour drugs, particularly hydroxyurea which showed increase in the production of HbF in non-human primates and patients with SCA [61-63].
Chemical inhibition of Hb polymerization: The controversy over the mechanism of inhibition of HbF by 5-Azacytidine laid the groundwork for a study in which hydroxyurea, a relatively non-toxic, S-phase-specific chemotherapeutic agent with readily reversible myelosuppressive effects which does inhibit the enzyme DNA methyltransferase was shown to result in a marked increase in the levels of HbF in baboons [64].
Hydroxyurea, an inhibitor of ribonucleotide reductase that was in use for several years for the treatment of myeloproliferative disorders is an orally available drug, relatively well tolerated and easy to use. After demonstration of its ability to induce the production of increased levels of HbF in baboons, a number of clinical trials were carried out in patients with SCA who were administered doses sufficient to cause mild myelosuppression. These patients showed marked increase in the number of HbF–containing red cells and the percentage of HbF in addition to the level of HbF per F cell [62,65]. In a separate national multicentre study comprising 299 adults with moderate to severe SCA, it was observed that the drug was relatively nontoxic and showed a marked decrease in the frequency and severity of painful crises. In addition to this, considerable reduction in the incidence of acute chest syndromes, the need for transfusions and hospitalizations were also observed [66,67]. Additional studies in children with SCA demonstrated the clinical efficacy and short-term safety of hydroxyurea resulting in a substantial reduction of hospitalization [68-70]. Albeit, similarities were observed in the HbF inducing activity of hydroxyurea and 5-azacytidine among anaemic baboon and patients with SCA. However, its molecular targets and mechanisms of action require further elucidation. The original theory suggested that hydroxyurea may elevate HbF levels by accelerating the differentiation of erythroid formation in the bone-marrow, leading to the appearance of ‘foetal-like’ cells in the peripheral blood [71]. More conventional theories have shown that hydroxyurea generates NO in vivo resulting in the activation of the NO/cGMP signalling pathway leading to the up-regulation of γ-globin gene expression in patients with SCA [72]. Additionally, the hydroxyurea has other effects that may be beneficial to patients with SCA. For example, hydroxyurea has shown to reduce the number of reticulocytes and young, lowdensity sickled red cells to the endothelium and also decrease the expression level of soluble VCAM-1 [73,74].
Butyrate: Due to the concerns about the long-term administration of an antitumor drug and its potential serious side effects, there has been considerable interest in identifying safe and effective inducers for the production of HbF. Initially, studies on recombinant human erythropoietin suggested promising results in stimulating HbF production in patients with SCA [75]. However, subsequent studies on human erythropoietin administration unaided or in combination with hydroxyurea have yielded conflicting results [76-78]. This led to a considerable interest in the physiologic and pharmacologic roles of butyrate in the regulation of HbF production.
A short-chain fatty acid that inhibits HDAC was shown to stimulate embryonic or foetal gene expressions in sheep, mice, chicken and baboons [79-81]. Furthermore, the switch from HbF to HbA in infants with diabetic mothers is delayed due to the increased serum concentration of amino-n-butyric acid [82]. Additionally, studies involving intermittent administration of arginine butyrate in patients with SCA resulted in sustained induction in the production of HbF [83]. While there exists considerable evidence of this agent in the treatment of SCA, there is an added need to assess the efficacy and safety of the administration of this drug.
The butyrates are perhaps the first class of drugs designed to transcriptionally activate specific genes--in this particular case, to reactivate the developmentally silenced foetal globin genes. In recent times, research focusing on the conventional use of sodium butyrate is used for foetal haemoglobin induction is being studied. It is suggested that sodium butyrate treatment can be replaced by safer therapeutic tools like microRNAs, small non-coding RNAs that control number of epigenetic mechanisms. In a study by Tayabi and colleagues, it was demonstrated that gamma chain was 5.9-fold up-regulated in the treated group [84].
It can be assumed that sodium butyrate can up-regulate gamma chain gene, and change the expression of micro RNAs (miRNAs). These results can be profitable in future studies to discover therapeutics suitable for such disorders.
Bone marrow transplantation: The concept of replacing the source of defective sickled RBCs, the bone marrow, with a healthy bone marrow which produces normal RBC is an insightful therapeutic approach in the treatment of SCA. However, for several years the possibility of such an approach was considered uncertain for a non-malignant disorder like SCA, due to the high chances of mortality. Nonetheless, recent advancements in immunosuppressive therapy and supportive care resulted in reduction in the mortality rates following bone marrow transplantation (BMT). Patients with β-thalassemia, after treatment with BMT showed a considerable 90% increase in survival rates [85]. Promising results such as those described above led to improved interest in the use of BMT as a therapy for treatment of SCA and clinical trials were conducted in Europe and USA to exploit the full potential of this therapy [86-88]. However, the foremost drawback in using BMT as a therapeutic approach lies in the fact that it is almost impossible to find a matched sibling donor as suitable candidate for transplantation [89]. Nevertheless, efforts and clinical trials are underway to increase the availability of sources of haematopoietic stem cells (HSCs) for transplantation to identify procedures to reduce the mortality and morbidity rates in adults [90]. Currently, BMT appears to be the only effective curative theory and the main challenge that exists is to make it commonly available to patients.
Future Prospects
Gene therapy
In order to cure or prevent SCD, researchers could settle patient’s undifferentiated cells of hemoglobin by utilizing quality altering property and afterward transplant the changed cells back into the patient. In the event that specialists could alter a patient's hematopoietic undifferentiated cells, either to settle the β-globin transformation or to restart the generation of fetal hemoglobin. Thereafter, utilize them to repopulate the bone marrow. As we know that, there is rare compelling reason to locate a good bone-marrow contributor.
Repairing or replacing the β-globin gene is not a new idea, but researchers have found fresh promise in the relative ease of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technique, achieving early success in mouse models and genome editing system in the treatment of cancer [91]. The materials required for CRISPR are easily accessible. Researchers need only an enzyme that cuts DNA, such as the endonuclease Cas9, and a guide RNA molecule that directs the enzyme to cut the gene of interest. Instead of leaving the cell to make its own error-prone repairs to the CRISPR-induced cuts, researchers supply further guidance by delivering a strand of DNA that contains the corrected snippet of the haemoglobin sequence- a kind of ‘repeat after me’. This approach is called homologydirected repair [84].
Repairing or supplanting the β-globin quality isn't another thought, yet researchers have discovered new guarantee in the relative simplicity of the CRISPR method, making early progress in mouse models. CRISPR-Cas9 was adjusted from a normally happening genome altering framework in microbes. The microscopic organisms catch pieces of DNA from attacking infections and utilize them to make DNA fragments known as CRISPR exhibits. The CRISPR exhibits enable the microbes to "recollect" the infections (or firmly related ones). In the event that the infections assault once more, the microorganisms create RNA sections from the CRISPR clusters to focus on the infectious' DNA. The microscopic organisms at that point utilize Cas9 or a comparable catalyst to cut the DNA separated, which handicaps the infection [92]. The materials required for CRISPR are effortlessly open. Analysts require just a chemical that cleaves DNA, for example, the endonuclease Cas9, and a guide RNA molecule which guides the protein to cut the quality of intrigue. Rather than leaving the gene to make its own particular blunder inclined repairs to the CRISPR-instigated cuts, specialists supply facilitate direction by conveying a strand of DNA that contains the remedied piece of the haemoglobin succession- a sort of 'rehash after me'. This approach is called homologycoordinated repair.
Red blood cells produced from the edited haematopoietic stem cells of patients with SCD produced 25% more foetal haemoglobin than unedited cells, and the edited cells successfully engrafted in mice. After those cells are transplanted into mice with smothered invulnerable frameworks, their numbers tumble off: 10% of the human cells that were joined, or engrafted, into the bone marrow of the mice created solid haemoglobin. Future research can be steered in this direction, where alteration in various factors can attain the results [93]. Another study performed in patients with SCD who received gene therapy with the use of lentiviral gene addition of an antisickling β-globin variant, showed that normal blood cell counts were attained after the transduced stem cells were engrafted. While adverse events related to the BB305-transduced cells were not observed, and the pattern of vector integration remained polyclonal without clonal dominance, insertional oncogenesis was reported in clinical gene-transfer studies with gamma retroviral vectors [94,95].
This successful experiment suggests that SCD can be cured by using gene therapy technique; however, it is not implemented in regular patients of SCA due to the fact that it has to be tested on a large scale. In this regard, it opens the door to further research by considering a large population to see its success.
Conclusion
International health care providers and health care agencies are faced with multiple challenges to overcome the burden of SCA among children in backward countries. In order to spearhead the process, emphasis should be laid on diagnostic procedures such as new-born screening, and anti-microbial prophylaxis. While these are affordable resources available in low income countries, efforts should also be made to improve morbidity as well as quality of life. Many advances have taken place in the past decade to improve treatment options for SCA. However, most of these treatments are expensive and dependent on medical infrastructure and only a few patients with SCA are able to afford treatments such as gene therapy and hematopoietic stem cell transplantation. There is a need for active measures and collaborations to be deployed on the part of the government to effectively implement population screening. Additionally, counseling programs and epidemiological studies in SCA stricken countries are necessary to assess current and future health needs.
22306
References
- Matos A Malheiros C Matos S (2016) Health related quality of life of patients with sickle cell disease. J Blood Disord Trans 7: 1-3.
- Weatherall D Hofman K Rodgers G Ruffin J Hrynkow S (2005) A case for developing north-south partnerships for research in sickle cell disease. Blood 105: 921-923.
- Herrick J (1910) Peculiar elongated and sickle shaped red blood corpuscules in a case of severe anemia. Arch Int Med 6: 517-521.
- Pauling L Itano HA Singer SJ Wells AC (1949) Sickle cell anemia: A molecular disease. Science 110: 543-549.
- Ingram V (1956) A specific chemical difference between the globins of normal human and sickle-cell anemia haemoglobin. Nature 178: 792-794.
- Ashley-Koch A Yang Q Olney RS (2000) Sickle hemoglobin (Hb S) allele and sickle cell disease: A HuGE review. Am J Epidemiol 151: 839-845.
- Wang Z Qiu C Wang S Han Z Huang Z et al. (2014) Effect of hemoglobinA1C on the coronary flow velocity after percutaneous coronary intervention. Biomed Res 26: 214-218.
- Wang P Guan H Wang ZM Xu CH (2014) Clinical evaluation of intra-arterial thrombolysis treatment of wake-up stroke under CT perfusion imaging guidance. Biomed Res 26: 135-141.
- Flint J Harding R Boyce A Clegg J (1998) The population genetics of the haemoglobinopathies. Baillieres Clin Haemato 11: 1-51.
- Anie K Paintsi V Owusu-Dabo E Ansong D Osei-Akoto A et al. (2017) Organ damage in sickle cell disease study (ORDISS): Protocol for a longitudinal cohort study based in Ghana. BMJ open 7: 1-7.
- Muoghalu CA Awolowo O (2017) The health workers perspectives in the management of sickle cell disease in an urban health centre in ile-ife Nigeria. J Hematol Thrombo Dis 5: 262.
- J. Neel (1949) The inheritance of sickle cell anemia. Science 110: 64-66.
- Weatherall D Akinyanju O Fucharoen S Olivieri N Musgrove P (2006) Inherited disorders of hemoglobin Disease control priorities in developing countries (2nd edition). New York: Oxford University Press.
- LonerganGJ Cline DB Abbondanzo SL (2001) Sickle Cell Anemia. RadioGraphics 21: 971-994.
- Rucknagel DL (1974) The Genetics of Sickle Cell Anemia and Related Syndromes. Arch Int Med 133: 595-606.
- Taliaferro WH Huck J (1923) The inheritance of sickle-cell anaemia in man. Genetics 8: 594-598.
- Epstein FH (1997) Pathogenesis and treatnment of sickle cell disease. N Engl J Med 337: 762-769.
- Odievre MH, Verger E, Silva-Pinto AC, Elion J (2011) Pathophysiological insights in sickle cell disease. Indian J Med Res 134: 532-537.
- Ali SD, George PD, Anbalagan M (2015) Impact of G406S and G420R mutants associated with blood coagulation factor Xa: Molecular simulation approach. Biomed Res 26: 682-685.
- Bunn HF (1999) Induction of fetal hemoglobin in sickle cell disease. Blood 93: 1787-1789.
- Bookchin RM, Lew VL (1996) Pathophysiology of sickle cell anemia. Haematol/Oncol Clinics North Am 10: 1241-1253.
- Rodgers GP (1997) Overview of pathophysiology and rationale for treatment of sickle cell anemia. Semin Hematol 34: 2-7.
- Aydogan MS, Ucar M, Yucel A, Karakas B, Gok A, et al. (2016) The effects of iron deficiency on red blood cell transfusion requirements in non-bleeding critically ill patients. Biomed Res 27.
- Blaisdell C, Goodman S, Clark K, Casella J, Loughlin G (2000) Pulse oximetry is a poor predictor of hypoxemia in stable children with sickle cell disease. Arch Pediatr Adolesc Med 154: 900-903.
- Sun K, Xia Y (2013) New insights into sickle cell disease: A disease of hypoxia. Curr Opin Hematol 20: 215-221.
- Andhale RB, Lodha S, Wankhade V (2013) renal physiological status of sickle cell anemic patients, district amravati, MS India. J Blood Disord Transfus 5: 188.
- Ortiz FO, Aldrich TK, Nagel RL, Benjamin LJ (1999) Accuracy of pulse oximetry in sickle cell disease. Am J Respir Crit Care Med 159: 447-451.
- Dickerhoff R (2002) Splenic sequestration in patients with sickle cell disease. Klin Padiatr 214: 70-73.
- Powars D (1990) Sickle cell anemia and major organ failure. Hemoglobin 14: 573-598.
- Sickle cell disease: Symptoms, diagnosis, treatment and recent developments (2018). NIH MEdline Plus 2011.
- Gallo A, Wilkie D, Suarez M, Labotka R, Molokie R, et al. (2010) Reproductive decisions in people with sickle cell disease or sickle cell trait. West J Nurs Res 32: 1073-1090.
- Devkota BP (2018) Hemoglobin Electrophoresis. Drugs & Diseases, Laboratory Medicine.
- Adjei GO, Goka BQ, Enweronu-Laryea CC, Rodrigues OP, Renner L, et al. (2014) A randomized trial of artesunate-amodiaquine versus artemether-lumefantrine in ghanaian paediatric sickle cell and non-sickle cell disease patients with acute uncomplicated malaria. Malaria J 13: 369.
- Parthasarathi D, Gajendra C, Dattatreya A, Sree Venkatesh Y (2011) Analysis of Pharmacokinetic & Pharmacodynamic Models in Oral and Transdermal Dosage Forms. J Bioequiv Availab 3: 268-276.
- Eke F (2005) An open comparative study of dispersible piroxicam versus soluble. Trop Med Int Health 5: 81-84.
- Beyer JE (2000) Judging the effectiveness of analgesia for children and adolescents during vaso-occlusive events of sickle cell disease. J Pain Sympt Managet 19: 63-72.
- Richardson P (1993) Meperidine and ketorolac in the treatment of painful sickle cell crisis. Annal Emerg Med 22: 1639-1640.
- Goodman E (1991) Use of ketorolac in sickle cell disease and vaso-occlusive crisis. Lancet 338: 641-642.
- Majrashi AH, Alsaeed AH, Alsaeed AM, Shaik AP (2016) Correlation of haemoglobin-f levels with biochemical parameters in pediatric patients with sickle cell anemia from Jazan Saudi Arabia. Biomed Res 27: 702-709.
- Strouse J, Heeney MM (2012) Hydroxyurea for the Treatment of Sickle Cell Disease: Efficacy Barriers Toxicity and Management in Children. Pediatric Blood Cancer 59: 365-371.
- Hydroxyurea Treatment for Sickle Cell Disease (2008) St. Jude Children’s Research Hospital.
- Queiroz AMM, de Castro Lobo CL, do Nascimento EM, de Bragança Pereira B, Bonini-Domingos, et al. (2013) The Mean Corpuscular Volume and Hydroxyurea in Brazilian Patients with Sickle Cell Anemia: A Surrogate Marker of Compliance. J Blood Disord Transfus 4: 157.
- T. Wong, Brandow A, Lim L (2014) Update on the use of hydroxyurea therapy in sickle cell disease. Blood 124: 3850-3857.
- Ballas S, Singh P, Adams-Graves W (2013) Idiosyncratic side effects of hydroxyurea in patients with sickle cell anemia. J Blood Disord Transfus 4: 1-3.
- Beverung L, Brousseau D, Hoffmann R, Yan K, Panepinto J (2014) ambulatory quality indicators to prevent infection in sickle cell disease. Am J Hematol 89: 256-260.
- Nagel RL, Bookchin R, Johnson J, Labie D, Wajcman H, et al. (1979) Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci USA 76: 670-672.
- Perrine R, Pembrey M, John P, Perrine SF (1978) Natural history of sickle cell anemia in Saudi Arabia. A study of 270 subjects. Ann Internal Med 88: 1-6.
- Kar BC (1986) Sickle cell disease in orissa state, india. Lancet 2: 1198-1201.
- Atweh GF, Frenette PS (2007) Sickle cell disease: Old discoveries, new concepts and future promise. J Clinl Investigat 117: 850-858.
- Lawn R, Fritsch E, Parker R, Blake MT (1978) The isolation and characterization of linked delta- and beta-globin genes from a cloned library of human DNA. Cell 15: 1157-1174.
- Collins FS, Weissman SM (1984) The molecular genetics of human haemoglobin. Prog Nucleic Acid Res Mol Biol 31: 315-462.
- Orkin S (1990) Globin gene regulation and switching: Circa 1990. Cell 63: 665-672.
- Groudine M, Weintraub H (1981) Activation of globin genes during chicken development. Cell 24: 393-401.
- van der Ploeg LH, Flavell RA (1980) DNA methylation in the human gamma delta beta-globin locus in erythroid and nonerythroid tissues. Cell 19: 947-958.
- Mavilio F, Giampaolo A, Care A, Migliaccio G, Calandrini M, et al.(1983) Molecular mechanisms of human hemoglobin switching: Selective undermethylation and expression of globin genes in embryonic fetal and adult erythroblasts. Proc Natl Acad Sci USA 80: 6907-6911.
- DeSimone J, Heller P, Hall ZL (1982) 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Natl Acad Sci 79: 4428-4431.
- Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, et al. (1984) Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Investigat 74: 652-656.
- Dover G, Humphries R, Moore J, Ley T, Young N, et al. (1986) Hydroxyurea induction of hemoglobin F production in sickle cell disease: Relationship between cytotoxicity and F cell production. Blood 67: 35-38.
- Charache S, Dover G, Moyer A, Moore W (1987) Hydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anaemia. Blood 69: 109-116.
- Letvin N, Linch D, Beardsley G, McIntyre D (1984) Augmentation of fetal-hemoglobin production in anemic monkeys by hydroxyurea. New J Eng Med 310: 869-873.
- Orringer E, Blythe D, Johnson A, Dover G, Parker J (1991) Effects of hydroxyurea on hemoglobin F and water content in the red blood cells of dogs and of patients with sickle cell anemia. Blood 78: 212-216.
- Charache S, Terrin M, Moore R, Dover G, Barton F, et al. (1995) Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. New Eng J Med 332: 1317-1322.
- Charache S, Barton F, Moore R, Terrin M, Steinberg M, et al. (1996) Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine Baltimore 75: 300-326.
- Papadopoulou E, Teli A, Theodoridou S, Gompakis M ( 2015) Safety and efficacy of hydroxyurea in children and adolescents with sickle/beta-thalassemia: Two-year experience. Hippokratia 19: 172-175.
- Hankins J, Ware R, Rogers Z, Wynn L, Lane P, et al. (2005) Long-term hydroxyurea therapy for infants with sickle cell anemia: The HUSOFT extension study. Blood 106: 2269-2275.
- Ferster A, Vermylen C, Cornu G, Buyse M, Corazza F, et al. (1996) Hydroxyurea for treatment of severe sickle cell anemia: A pediatric clinical trial. Blood 88: 1960-1964.
- Stamatoyannopoulos G (2005) Control of globin gene expression during development and erythroid differentiation. Experiment Hematol 33: 259-271.
- Cokic V, Smith R, Beleslin-Cokic B, Njoroge J, Miller J, et al. (2003) Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Investigat 111: 231-239.
- Saleh A, Hillen D (1999) Levels of endothelial neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematologica 102: 31-37.
- Bridges K Barabino G Brugnara C Cho M Christoph G et al. (1996) A Multiparameter Analysis of Sickle Erythrocytes in Patients Undergoing Hydroxyurea Therapy. Blood 88: 4701-4710.
- al-Khatti A Umemura T Clow J Abels RI Vance J et al. (1998) Erythropoietin stimulates F-reticulocyte formation in sickle cell anemia. Trans Assoc Am Physicians 101: 54-61.
- Rodgers GP Dover GJ Uyesaka N Noguchi CT Schechter AN et al. (1993) Augmentation by erythropoietin of the fetal-hemoglobin response to hydroxyurea in sickle cell disease. New Eng J Med 328: 73-80.
- Nagel RL Vichinsky E Shah M Johnson R Spadacino E et al. (1993) F reticulocyte response in sickle cell anemia treated with recombinant human erythropoietin: A double-blind study. Blood 81: 9-14.
- Goldberg MA Brugnara C Dover GJ Schapira L Charache S et al. (1990) Treatment of sickle cell anemia with hydroxyurea and erythropoietin. New Eng J Med 323: 366-372.
- Ginder GD Whitters MJ Pohlman JK (1984) Activation of a chicken embryonic globin gene in adult erythroid cells by 5-azacytidine and sodium butyrate. Proc Natl Acad Sci USA 81: 3954-3958.
- Constantoulakis P B Josephson Mangahas L Papayannopoulou T Enver T et al. (1991) Locus control region-A gamma transgenic mice: A new model for studying the induction of fetal hemoglobin in the adult. Blood 77: 1326-1333.
- Constantoulakis P G Knitter G Stamatoyannopoulos (1989) On the induction of fetal hemoglobin by butyrates: In vivo and in vitro studies with sodium butyrate and comparison of combination treatments with 5-AzaC and AraC Blood 74: 1963-1971.
- Perrine S Greene M Faller D (1985) Delay in the fetal globin switch in infants of diabetic mothers. New Eng J Med 312: 334-338.
- Atweh G Sutton M Nassif I Boosalis V Dover G et al. (1995) Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 93: 1790-1797.
- Tayebi B Abrishami F Alizadeh S Minayi N Mohammadian M et al. (2015) Modulation of microRNAs expression in hematopoietic stem cells treated with sodium butyrate in inducing fetal hemoglobin expression. Artif Cells Nanomed Biotechnol 45: 146-156.
- Lucarelli G Galimberti M Polchi P Angelucci E Baronciani D et al. (1991) Bone marrow transplantation in thalassemia. Hematol Oncol Clin North Am 5: 549-556.
- Vermylen C Cornu G Ferster A Ninane J Sariban E (1993) Bone marrow transplantation in sickle cell disease: The Belgian experience. Bone Marrow Transplant 12: 116-117.
- Bernaudin F Souillet G Vannier J Plouvier E Lemerle S et al. (1993) Bone marrow transplantation BMT) in 14 children with severe sickle cell disease (SCD): The French experience. Bone Marrow Transplant 12: 118-121.
- Walters MC1 Patience M Leisenring W Eckman JR Scott JP et al. (1996) Bone marrow transplantation for sickle cell disease. New Eng J Med 335 pp. 369-376.
- Walters MC Patience M Leisenring W Eckman JR Buchanan GR et al. (1996) Barriers to bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant 2: 100-104.
- Locatelli F Rocha V Reed W Bernaudin F Ertem M et al. (2003) Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood 101: 2137-2143.
- Bhattacharjee R Purkayastha K Adapa D Choudhury A (2017) CRISPR/Cas9 genome editing system in the diagnosis and treatment of cancer. J RNAi Gene Silencing 13: 585-591.
- Gupta RM Musunuru K(2014) Expanding the genetic editing tool kit: ZFNs TALENs and CRISPR-Cas9. J Clin Invest 124: 4154-4161.
- Bourzac K (2017) Gene therapy: Erasing sickle-cell disease. Nature 549: S29.
- Ribeil J Hacein-Bey-Abina S Payen E Magnani A Semeraro M et al. (2017) Gene therapy in a patient with sickle cell disease. New Eng J Med 376: 848-855.
- Zanoni CT Galvão F Junior AC Saad STO (2015) Is Aquatic Physical Therapy a Feasible Treatment for Musculoskeletal Dysfunction of Sickle Cell Disease Patients?. J Yoga Phys Ther 5: 216.


