Ma. Guadalupe Treviño-Alanís1,2*, Sergio Salazar- Marioni2, Héctor Martínez-Menchaca2, y Gerardo Rivera-Silva1,2
1Departamento de Ciencias Básicas, Universidad de Monterrey, San Pedro Garza García, Nuevo León, México.
2Laboratorio de Ingeniería Tisular y Medicina Regenerativa, Universidad de Monterrey, San Pedro Garza García, Nuevo León, México.
- *Corresponding Author:
- Ma. Guadalupe Treviño
Laboratorio de Ingeniería Tisular y Medicina Regenerativa
Departamento de Ciencias Básicas, Escuela de Medicina, Universidad de Monterrey
Av. Morones Prieto 4500 Pte., 66238, San Pedro Garza García N.L., México
Tel: (81) 8215 1446
E-mail: maria.guadalupe.trevino@udem.edu.mx
Keywords
Klippel-Feil. Congenital malformation. Cervical fusion.
Introducción
Klippel y Fiel describieron por primera vez las características clínicas de este padecimiento en 1912. El síndrome de Klippel- Feil es un padecimiento de herencia autosómica dominante con penetrancia disminuida y expresión inestable, con una incidencia no precisada debido a su rareza y a que la mayoría de los pacientes son asintomáticos; el análisis cromosómico revela un cariotipo normal. Algunos casos son dominantes ligados al cromosoma X. Hay una herencia heterogénea genética asociado a elementos ambientales lo que revela la heterogeneidad del síndrome.[1]
Las malformaciones se deben a una alteración en la migración del tejido mesodérmico en el momento de la formación de los discos cervicales y otros órganos, entre la tercera y cuarta semana de desarrollo embrionario.[2] En la fusión cervical tipo I se muestra una ensambladura total de las vértebras cervicales hasta las superiores dorsales. En la tipo II se delimita a una o dos vértebras, generalmente acompañadas de fusión occipitoatlantoidea; es la más común. El tipo III se asocia la fusión cervical a una alteración análoga a nivel dorsal o lumbar, y en la tipo IV hay fusión cervical, torácica superior, dorsal inferior o lumbar.[3]
El diagnóstico puede sospecharse por la existencia de cuello corto y ancho, paladar hendido, implantación baja de cabello posterior y reducción de la movilidad del cuello.[4] Además, suele ser frecuente la asociación con malformaciones congénitas, entre ellas cardiopatías (4-14%), siendo la más frecuente la comunicación interventricular.[5] El diagnóstico se confirma mediante un examen radiológico que se caracteriza por fusiones de las vértebras cervicales y otras alteraciones óseas como discos ausentes o hipoplásicos, pérdida de altura del cuerpo vertebral y ocasionalmente hemivértebra.[6]
El tratamiento y pronóstico son variables de acuerdo al grado de compromiso de las funciones del paciente, van a depender de los problemas neurológicos asociados y del tipo de cardiopatía relacionada y el tiempo de evolución que influye en la presentación de complicaciones. Cada caso debe ser analizado para decidir el manejo médico o quirúrgico.
Caso clínico
Mujer de 26 años de edad con antecedente de hipotiroidismo diagnosticado y tratado hace dos años. Acudió a consulta por secreción mucosa retronasal y problemas para deglutir, traía el diagnóstico de hernia hiatal. En la exploración física presentó implantación baja del cabello posterior, cuello corto y cierta limitación de la movilidad del cuello. En el esofagograma se observó paso rápido del material de contraste, sin evidencia de compresión intrínseca o extrínseca ni otra anormalidad que comprometiera el paso de contraste a través del esófago. Ante la sospecha diagnóstica del síndrome de Klippel-Feil, se realizó una radiografía simple lateral de cuello y se identificó fusión de cuerpos vertebrales y arcos posteriores cervicales a nivel de C5-C6 (Figura 1). Para ratificar el diagnóstico, se efectuó unatomografía computada (TC) de cuello en la que se encontró el hallazgo característico del síndrome de Klippel-Feil, la fusión de vértebras cervicales; además se realizó una TC de cráneo donde se identificó una celdilla de Haller izquierda como variante anatómica (Figura 2). El análisis del caso fue completado al descartarse malformaciones cardiacas por ecocardiografía; pulmonares, nerviosas y renales por TC.
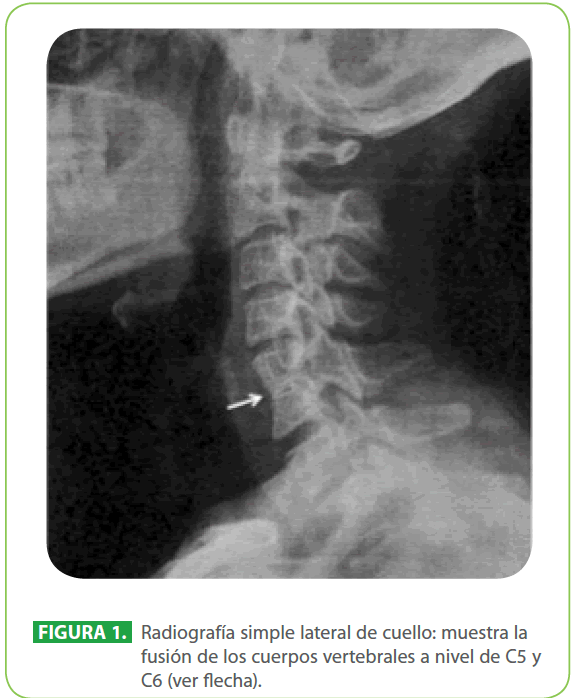
Figura 1: Radiografía simple lateral de cuello: muestra la fusión de los cuerpos vertebrales a nivel de C5 y C6 (ver flecha).
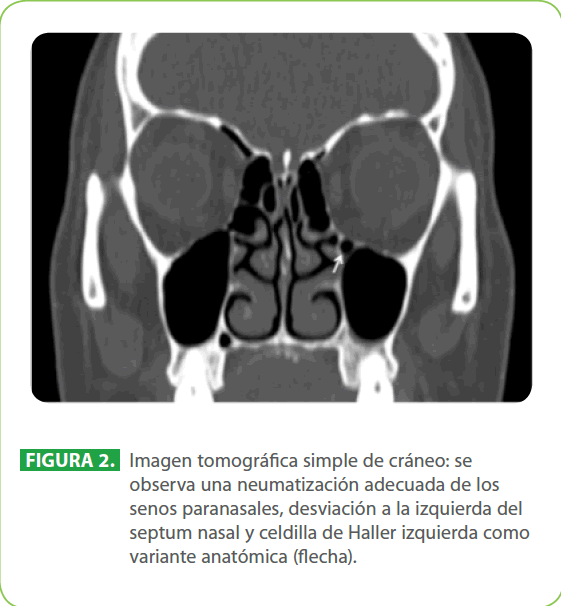
Figura 2: Imagen tomográfica simple de cráneo: se observa una neumatización adecuada de los senos paranasales, desviación a la izquierda del septum nasal y celdilla de Haller izquierda como variante anatómica (flecha).
Discusión
Aunque es poco habitual, este síndrome puede estar relacionado con otras alteraciones músculo-esqueléticas como artrodesis posterior, siringomelia, escoliosis o cifosis, tortícolis por contractura muscular, deformidad de Sprengel (escápula alta por ausencia de migración durante la gestación), hipoplasia unilateral del músculo pectoral mayor, sindactilia, dedos supernumerarios, entre otros. [6-8] Este padecimiento puede estar coligado con graves alteraciones congénitas como riñón en herradura, agenesia renal, criptorquidia, ausencia de vagina y ovarios, duplicación intestinal, hemiparesia, quiste broncogénico, ptosis, encefalocele, hidrocefalia, dolor de cuello, sordera, coloboma, comunicación interventricular, entre otras. [9] Se han comprobado deleciones genéticas en 5q 11.2 y 8q 22.2, en familias con síndromes genéticos que incluían la anomalía de Klippe-Feil. [10] La sospecha diagnostica es clínica, y posteriormente el diagnóstico es confirmado mediante radiografía simple. La ecocardiografía, la TC y la imagen por resonancia magnética, son métodos auxiliares para descartar la asociación con de otras anomalías congénitas. El síndrome de Klippel-Feil no tiene un tratamiento específico, debiendo descubrirse todas las malformaciones asociadas con la finalidad de tratarlas cuando sea factible.
739
References
- Tracy MR, Dormans JP, Kusumi K. KlippelFeil syndrome: clinical features and current understanding of etiology. ClOrthop Relate Res 2004;424:183-190.
- Allsopp GM, griffiths S, Sgouros S. Cervical disc prolapse in childhood associated with Klippel-Fiel syndrome. Childs NervSyst 2001;17:69-70.
- Franzen D, Sculte B, Beyer D, Neidel J, Koebke J, de Vivie R. Klippel-Fiel syndrome associated with aortic coarctation. Cardiovascular Pathol 2003;12:115-117.
- Hikade KR, Bitar GJ, Edgerton MT, Morgan RF. Modified 2-plasty repair of webbed neck deformity seen in Turner and Klippel-Fiel syndrome. Cleft Palate Craniofac J 2002;39:261-266.
- Hinojosa M, Tatagiba M, Harada K, Samii M. Dermoid cyst in the posterior fossa accompanied by Klippel-Fiel syndrome. Child NervSust 2001;17:97-100.
- Smith BA, Griffin C. Klippel-Feil syndrome. Ann Emerg Med 1992; 21: 876-879.
- Stallmer ML, Vanaharam V, Mashour GA. Congenital cervical spine fusion and airway management: a case series of Klippe-Feil syndrome. J ClinAnesth 2008, 20: 447-451.
- Franzen D, Schulte B, Beyer D, Neidel J, Koebke J, Vivie R. Klippel- Feil syndrome associated with aortic coarctation. Cardiovascular Pathology 2003; 12: 115-117.
- Alter AH. Klippel-Feil syndrome: A constellation of associated anomalies: R.N. Hensinger, J.E. Lang, and MacEwen. J. J PediatrSurg 1975; 10: 288-289.
- O‘Donnel DP, Seupaul RA. Klippe-Feil syndrome. Amer J Emerg Med 2008; 26: 257-261.

